Способ получения замещенных 1н-имидазолов или их солей присоединения нетоксичных, фармацевтически приемлемых кислот
Похожие патенты | МПК / Метки | Текст | Заявка | Код ссылки














Текст
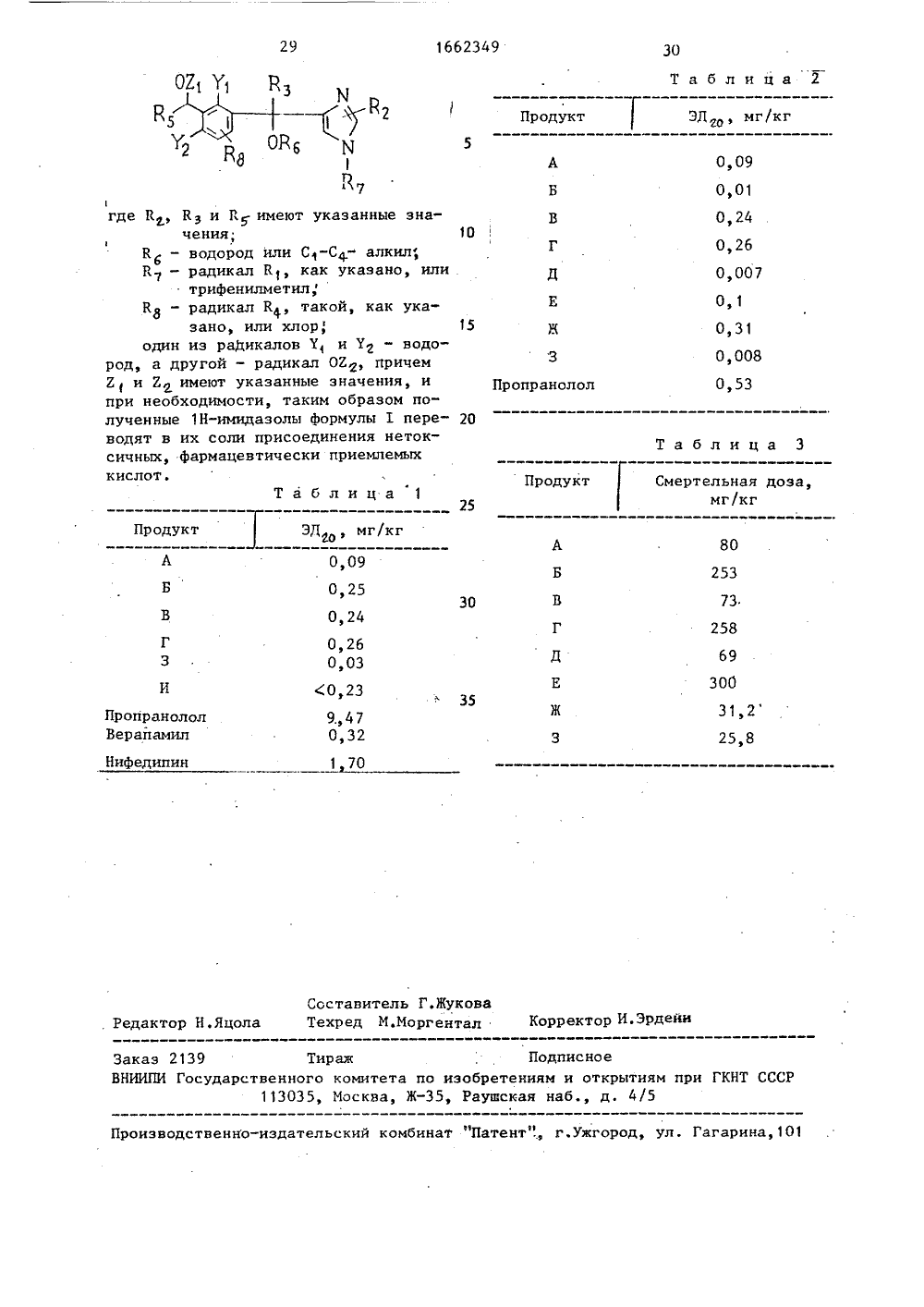
СОЮЗ СОВЕТСКИХСОЦИАЛИСТИЧЕСКИХРЕСПУБЛИК ГОСУДАРСТВЕННЫЙ КОМИТЕТ ПО ИЗОБРЕТЕНИЯМ И ОТНРЬГИЯ И ГКНТ СССР М ОЛИСАНИЕ ИЗОБРЕТЕНИЯНТУ ков астностиимидазолов В ля эт дящих препараты, примен е целей. я к способуных 1 Н-имидаоединения нески приемлеут быть исз таких свойств являетСовокупнос ся не характе;дазольного ря нен но для со ия - синтез новых льного ряда, обламическими, сердечны- и тканевыми свойствойствам превосхоПрименых соединениКт - алкил и 1. Получение ормулы 11 (К С или трифе с Н,илме тил) .(54) СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 1 НИКЩАЗОЛОВ ИЛИ ИХ СОЛЕЙ ПРИСОЕДИНЕНИЯНЕТОКСИЧНЫХ, ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ КИСЛОТ(57) Изобретение относится к гетероциклическим соединениям, в чк получению замещенных 1 Нф-лы Изобретение относит получения новых замеще олов или их солей при токсичных, Фармацевтич мых кислот, которые мо пользованы в медицине. Цель изобретен соединений имидазо дающих противоише ми, церебральными в , и по своим с)5 С 07 0 233/58//А 61 К 31/4 5 где К, К, К и К могут быть одинаыми ипй разными и каждый Н или С-Со-алки ьн радикалу К 4 Ну С-С,.-алкильный радикал или С-С-алкоксильный радикал, один иэ Уи Уд - Н, а другой - радикал 02, 2 и 2 взятые по отдельности, -С-С-алкильный радикал, а вместе обозначают группу -СН- или -С(СНэ), или их солей присоединения нетоксичных, фармацевтически приемлемых кислот, обладающих противоишемическими, сердечными, церебральными и тканевыми свойствами. Цель - разработка способа получения указанных соединений. Получают восстановлением имидазольного соеди- се нения и при необходимости переводом в их соли присоединения нетоксичных, фармацевтически приемлемых кислот.3 табл.кантируют, сушат.над сульфатом натрия, затем перегоняют. Остаток, который содержит трифенилметан, частичнорастворяют в 50 мл воды, содержащей3,3 мл концентрированной соляной кислоты, и полученную водную суспензиюэкстрагируют эфиром. Водную фазу затем доводят до рН 8 добавлением бикарбоната натрия, после чего экстрагируют дихлорметаном. Затем растворитель удаляют путем отгонки и получают 3,5 г О-(2,2-диметилН,3-бензодиокси-ил)-1 Н-имидазол-метанола, Выход ЗЗЙ.15Продукт образует хлоргидрат (перекристаллизуемый из смеси этанола сэфиром 1:1), который находится в стеклообраэном виде и не имеет четкой точки плавления.Вычислено, 7: С 56,66, Н 5,40,Н 9,44, С 1 11,97.С(4 Н 600 э НС 1Найдено, 7.: С 55,81 Н 57,И 8,94, С 1 11,83.б. ь-(2,6-Диметокси-З-метокснметилфенил)-1 Н-имидазол-метанол.Поступают как в п.1, но при 80 С,исходя из б-(2,6-днметокси-З-метоксиметилфенил)-1-трифенилметилН-имида 30зол-метанола (получен в примере1,В,8). После удаления выкристаллизовавшегося трифенилметана и выпаривания метанола полученный остаток очищают путем хроматографии на диоксидекремния (элюирующее средство: смесь 35дихлорметана с метанолом 80:20). Получают масло, охарактеризованное егоЯНР-спектром. Выход 683,ЯИР-спектр (СРС 1), 8: 3,38 (ЗН, 40с., ОСН), 366 (ЗН, с., ОСН), 3,76(тетрагидрофуран-эфир),Вычислено, Е: С 61,64, Н 6,85,Н 9,59,С Нго 02.04 55Найдено, 3: С 61,48, Н 7,0,М 9,38.8. Ж-н-Бутил-(2,2-диметилН 1,3-бензодиоксин-ил)-1 Н-имидаэол- е 4-метанол. Поступают как в п.1 но при 80 С исходя изО-н-бутил"е-(6-хлор,2-диметилН,3-бензодиоксин-ил)- 1-трифенилметилН-имидаэол"метанола (получен в примере 1,В,11). Остаток, полученный после фильтрации трифенилметана и выпаривания метанола, используют в следующей стадии.9.- Хлоргидрат о-(2,2-диметилН,3-бензодиоксин-ил)-0;метилН- имидазол-метанол. Поступают как в п.2, но при 20 С, исходя из о-(6- хлор,2-диметилН,3-бензодиоксин-ил)"6-метил-трифенилметил Н-имидазол-метанола (получен в примере 2,Б,2). Выход почти количественный. Т.пл. 85-100 С (разложение).Аналитический образец получают путем перемешивания вдиэтиловом эфире. Т.пл. 72-90 оС (разложение).Вычислено, 7: С 57,97, Н 6,12, Н 9,01, С 1 11,43С 11 щ М 20 з НС 1Найдено, Ж; С 57,94, Н 6,95, В 8,12, С 1 951.П р и м е р 4. Получение 1 Н-имидаэолов Формулы 1 (Е= Е = глкил и Е 1+ 2 = -СН-).1. Хлоргидрат 4-(4 Н,3-бензодиоксин-ил)-метилН-имидазола, Подвергают гидрогенолизу 50,85 г (0,1 моль) 0-(б-хлорН,3-бензодиоксин-ил)-1-трифенилметилН-имидазол-метанола (получен в примере 1,В., 1)растворенного в 500 мл уксусной кислоты, в присутствии 3 г 107- ного палладия на угле, в течение 2 чопри 80 С под давлением водорода 2,41 бар, затем катализатор удаляют фильтровыванием и растворитель отгоняют при пониженном давлении. Полученный остаток экстрагируют три раза диэтиловым эфиром для удаления триФенилметана, который образовался. Его перекристаллиэуют затем из 50 мл ацетонитрила. Получают 21,2 г хлоргидрата 4-(4 И,3-бензодиоксин- ил)-метилсяН-имидазола. Выход 83%, т,пл, 168-173 ОС.Вычислено, 7.; С 57,03, Н 5,15, М 11,09, С 1 14,06.СНИО НС 1Найдено, Х: С 57,01, Н 5,20, И 11,02, С 1 13,83.2. 4-(4 Н,3-Бензодиоксин-б-ил)метилН-имидазол. Это соединениеполучают как и предыдущее соединение, 21 1662349но из 25 г (0,052 моль) е - (43-1,3 бензодиоксин-ил)-1-трифенилметил 1 Н-имидазол-метанола (получен впримере 1,В 2). Образовавшийся трифе 1 Э ф5нилметан экстрагируют несколько разгексаном и остаток очищают хроматографией на диоксиде кремния (элюируюцее средство: дихлорметан и метанол95;5). фракции, содержащие продукт,,выпаривают и остаток обрабатываютразбавленным раствором аммиака в метаноле. Этот раствор перегоняют и остаток перекристаллизуют иэ этилацетата. Получают 3,5 г 4- (4 Н,3-бензодиоксин-ил)-метилсяН-имидазол. Выход 31,2%, т,пл. 145 С.Вычислено, %: С 66,66, Н 5,55,И 12,96,С(1 Н 12 ИЙ 01,Найдено, %: С 66,14, Н 5,62,Л 12,69.3, 4-(2,6-иметокси-З-метоксиметилфенил)-метилсяН-имидаэол-(хлергидрат) 251 20 В 1 л жидкого аммиака вводятб,75 г(0,0243 моль) 0-(2,6-диметокси-З-метоксиметилфенил)-1 Н-имидазол-метанола (получен в примере 3,6), предварительно растворенного в 200 мл тетрагидрофурана. Добавляют, во-первых,1,3 г (0,0243 моль) хлорида аммония,и затем, за 30 мин, 1,12 г (0,0486 моль)натрия кусочками. После полного исчезновения натрия добавляют 1,3 гхлорида аммония и перемешивают реакционную смесь в течение 30 мин. До,бавляют 300 мл толуола, содержащего10 мл метанола. Аммиак затем удаляютна водяной бане, Добавляют еще 100 мл 40воды и органическую фазу декантируют.Ее сушат над сульфатом натрия и растворитель выпаривают при пониженномдавлении. Полученный продукт очищаютс.помощью хроматографии на диоксиде 45кремния (элюирующее средство; смесьдихлорметана с метанолом и аммиаком95:4,5:0,5). Получают 1,81 г вещества в виде лака (выход 28,4%). Его растворяют в диизопропиловом эфире ипревращают в хлоргидрат путем добавления эквивалента раствора солянойкислоты и изопропиловом спирте. Т.пл.130-135 С.Вычислено, %: С 56,28, Н 6,36,И 9,38.С 14 Нз ИО з НС 1Найдено, %: С 56,13, Н 6,39,И 9,10. 4, 4-(2,6-Диметокси-З-(1-метоксиэтил)фенил)метилН-имидазол (хлоргидрат), Получают как в п.З, но исходят из К -2,6-диметокси-З-(1-метоксиэтил)-фенилН-имидазол-метанола(получен в примере 3,7). Полученноепосле выпаривания толуола масло обрабатывают эфиром и превращают в хлоргидрат путем добавления раствора соляной кислоты в метаноле. Выход68,5%, т.пл. 169-170 С,Вычислено, %: С 57,6, Н 6,72,Н 8,96, С 1 11,36.С Н о И О НС 1Найдено, %: С 57,52Н 6,84,Я 8,89, С 1 11,27.5. 4-1-(4 Н 3-Бензодиоксинил)-этилН-имидазол. Получают какв п.1но исходя из М-(б-хлорН,3 бензодиоксин-ил)-ф(;метил-трифенилметилН-имидаэол-метанола (получен в примере 2,Б, 1), После удаления трифенилметана, полученньй остаток растворяют в воде и раствор подщелачивают до рН 8 путем добавлениянасыщенного раствора карбоната натрия. Осадок, которьй, образуется,снова растворяют в дихлорметане.Органическую фазу сушат над сульфатом натрия и вываривают растворитель при пониженном давлении, Оста,. ток хроматографируют на диоксиде кремния (элюируюцее средство: смесь дихлорметана с метанолом и аммиаком 95:4,5:0,5), Выход 52%, т.пл. 145- 148 С (этилацетат).Вычислено, 7.: С 67,83, Н 6,09, М 12,17.Найдено, %: С 6768, Н 6,11,Б 12, 10. Таким образом, полученное соединение находится в рацемической форме.Его можно разделить на его два энантиомера следующим образом. При нагревании растворяют 7,57 г (0,0329 моль) рацемического 4-1-(4 Н,3-бенэодиоксин-ил)-этилН-имидазола в 263 мл изопропилового спирта. Туда добавляют 2,47 г (0,0165 моль). о-винной кислоты, растворенной в 33 мл изопропилового спирта. Образуется хлопьеобразный осадок. Суспензию перемешивают в течение 16 ч при комнатной температуре, затем удаляют жидкую фазу декантацией, Полученный остаток обрабатывают 300 мл кипящего изопропилового спирта. Нерастворив 1662349шаяся Фракция кристаллизуется. Охлаждают до 45 С и перемешивают еще смесьпри этой температуре в течение 4 ч.После этого фильтруют горячей (45 С).Получают 3,21 г кристаллической соли+40,80 (С = 1, метанол).Фильтрат, происходящий от отделения кристаллов, будет обрабатыватьсяв п.б).а) Полученные кристаллы сначалаперекристаллизуют два раза из изопропилового спирта, Т,пл. 178 С. (И)= +53,3 (с = 1, метанол).Затем 1 г этой соли растворяют в33 мл воды и подщелачивают раствор дорН 9,5 добавлением 1 н, раствора гидроксида натрия. Образуется осадок,который отфильтровывают и перекристаллизуют из 1 мл этилацетата. Такимобразом получают 0,457 г правовращающего изомера 6-4-1-(4 Н,3-бенэодиоксин-ил)-этилН-имидазола. Т.пл,113,7 С, (00 ) = +69,8 (с = 1, метанол) .Вычислено, %: С 67,83, Н 6,09,И 12, 17.С 1 Н 1 ИО,Найдено, %: С 67,88, Н 6,06,В 12,13..б) фильтрат выпаривают при пониженном давлении для удаления изопропилового спирта. Полученный остатокрастворяют в воде и раствор подщелачивают до рН 9,5 добавлением водного1 н. раствора гидроксида натрия. Высвободившееся основание осаждается,его отфильтровывают, затем обрабатывают в изопропиловом спирте полуэквивалентом 1-винной кислоты. В средеосаждается образовавшаяся соль, ОтФильтровывают и перекристаллизуют этусоль из изопропилового спирта. Т.пл.178 С. (00= -51,57 (с = 1, метанол).45Затем ее снова растворяют в водеи подщелачивают раствором до рН 9,5добавлением водного 1 н. раствора гид. роксида натрия. Образуется осадок,который отфильтровывают и перекристаллиэуют два раза из этилацетата,Получают 0,296 г левовращающего изомера 1-4-1-(4 Н-,1,3-бензодиоксинил)-этил -1 Н-имидазола. Т.пл; 114,4 С,(М)-72,6 (С 1, метанол).5. о ф 55Вычислено, %: С 67,83, Н 6,09,Н 12,17Сз Н 4 020 Найдено, %: С 67,78, Н 6,10,М 12,24.6. Хлоргидрат 4- (6-метилН,3 бензодиоксин-ил)-метилН-имидазола. Это соединение получают аналогичным образом. Т.пл. 175-183 С (разложение).Вычислено, %: С 58,58, Н 5,63,М 10,51С Н 14 ИОНС 1Найдено, %: С 56,47, Н 5,63,И 9,93.П р и м е р 5. Получение 4-1- .(2,2-диметилН,3-бензодиоксин(или.8)-ил)алкил -1 Н-имидазолов формулы 1 (2+" 2С(СН) ) е1. 4- (2,2-ДиметилН,3-бензодиоксин-ил)-метилН-имидазол. Подвергают гидрогенолизу 3,4 г (0,013 моль)ф-(2,2-диметилН,3-бенэодиоксинил)-1 Н-имидазол-метанола (получен впримере 3,4) в метаноле при 80 О-С втечение 5 ч в присутствии 10%-ногопалладия на угле и под давлением водорода 2,8 бар. Затем отфильтровываюткатализатор и растворитель удаляютпри пониженном давлении. Полученныйостаток хроматографируют на диоксидекремния (элюирующее средство: дихлбрметан;метанол 90;10). Получают 3 г4-(2,2-диметилН,3-бензодиоксин 6-ил)-метилН-имидазол. Выход 94%,т.пл. 150-170 С.ЯИР-спектр (ДЯСО), о: 1,45 (6 Н,с., СН-С-СН 3), 3,78 (2 Н, с., СН),4 77 (2 Н, с СН ), .6,5-7,7 (5 Н, м.,АгН + 1 шН),2 а. 4- (2,2-диметилН,3-бензодиоксин-нл)-кетилН-иыидееол.94, 13 г (0,137 моль) хлоргидратаф-(2,2-диметилН,3-бензодиоксин 8-ил)-1 Н-имидаэол-метанола (полученв примере 3, 1 или 3,5) вводят в 1 лбезводного жидкого аммиака. Дополняютрастворение реагента добавлением 1 лтетрагидрофурана. После этого добавляют кусочками 14,6 г натрия(0,634 моль). Спустя 5 мин после исчезновения натрия добавляют 34 г хлорида аммония и перемешивают реакционную смесь в течение 0,5 ч. Добавляют1,56 л 10%-ного раствора метанола втолуоле. Затем аммиак удаляют на водяной бане, После этого добавляют еще780 мл воды и отделяют органическуюфазу. Водную фазу экстрагируют двараза по 500 мл толуола. ОрганическиеФазы объединяют и промывают 1.л воды, 662349затем сушат над суль 4 атом натрия и перегоняют при пониженномдавлении. Полученный остатокперекристаллизуют из 900 мл1 кипящего ацетона. Таким образом получают 57,8 г 4-1(2,2-диметилН,3 бензодиоксин-ил)-метил 1-1 Н-имидазола, плавящегося при 160-170 С. Выход74,6 Х.Вычислено, Х: С 68,85, Н 6,56,Б 11,48.СПИ,О,.Найдено, Х: С 69,15, Н 6,70,М 11,53.2 б. Тот же самый продукт можетбыть получен из 0,8 г (0,0015 моль)К-(б-хлор,2-диметилН,3-бензодиоксин-ил)-1-трифенилметилН-имидаэол-метанола (получен в примере1,В,З) и 0,245 г (О 0106 коль) натриясогласно описанному в примере 3,5 способу. Полученный после выпариваниятолуольной Фазы остаток хроматографируют на 150 г диоксида кремния (10тут) (элюируюцее средство: этилацетат:метанол 95:5). Получают 123 мгпродукта, подобного выделенному вп.2 а.Следующие продукты получают способом, описанным в п.2 а.3. 4- (2,2-ДиметилН,3-бензодиоксин-ил)-метил -1-метилН-имидазол. Это соединение получают из хлоргидрата 0.-(2,2-диметилН,3-бензодиоксин-ил)-1-метилН-имидазолметанола (получен в примере 1,В, 12).Полученный остаток очищают с помощьюхроматографии на диоксиде кремния(диизопропиловый эфир-. гексан).Вычислено, Х: С 69,74, Н 7,02,И 10,84.С,Н,8 НО.Найдено, Х: С 69,93, Н 7,54,Б 10,78.4. 4-1(2,2-ДиметилН,3-бензодиоксин-ил)-метил -5-метилН-имидазол.Это соединение получают иэ 9,68 гсмеси хлоргидратов 0-метилированныхи спиртовых производных, полученнойсогласно примеру 3,2. Его очищают путем хроматографии на диоксиде кремния(элюирующее средство: дихлорметан-метанол 90: 10) . Получают 4 г целевогопродукта, Т.пл. 172-1780 С (дихлорметан) Вычислено, Х: С 69,74, Н 7,02,.Ю 10,84,С 1 Н 1 В Б 02.Найдено, Х: С 67,55, Н 6,98,Б 10,38.5. 4-(2,2,6-ТриметилН,3-бенэодиоксин-ил)-метил -1 Н-имидазол.Работают как в п.2 а, но исходя из2,3 г смеси, включающей 4-(2,2,6 триметилН,3-бенэодиоксин-ил)- (метокси)-метил -1 Н-имидазол, полученный в примере 3,3Остаток, полученный после выпаривания толуола, ис-.пользованного для экстракции продуктареакции, перекристаллизовывают иэ10 мл этилацетата. Выделяют 0,9 г целевого, продукта. Т.пл. 152-155 С(этилацетат).Вычислено, %: С 69,74, Н 7,02,20 Я 10 84.С Н 1 аигоаНайдено, Х: С 69,82, Н 7,04,И 10,64.6. 4-1-(2,2-ДиметилН,3-бензодиоксин-ил)этилаН-имидазол. Этосоединение получают из хлоргидратаф-(2, 2-диметилН,3-бензодиоксин 8-ил)метилН-имидазол-метанола(получен в примере 3,9) Полученныйпосле выпаривания толуола остатоккристаллизуется путем перемешиванияпри 600 С в диизопропиловом эфире. Выход 60 Х, т.пл. 118 и 130 С.. Вычислено, Х; С 69,74, Н 7,02,35 Я 10,84.СН 18 н,от.Найдено, Х; С 69,64, Н 6,95,Б 10,72,7. 4-1-(2,2-ДиметилН,3-бензодиоксиц-ил)-пентил -1 Н-имидазол,Это соединение получают восстановлением р-н-бутил-р-(2,2-диметилН,3 бензодиоксин-ил)-1 Н-имидазол-метанола (получили в примере 3,8). Выход 23,6 Х, т.пл. 108- 112 С (циклогексан).Вычислено, Х: С 72,0, Н 8,0,Н 9,33.С ц НгфИя.ОНайдено, Х: С 72,08, Н 8, 15,И 9,32. Фармакологическим испытаниям подвергались следующие продукты пред лагаемого способа:хлоргидрат 4-(б-метилН,3-бензодиоксин-ил)-метил,-1 Н-имидаэол (продукт А);16623427хлоргидрат 4-(4 Нр 3-бензодиоксин-ил)-метилН-имидазол (продукт Б);4- (2 р 2-диметилНр 3-бензодиоксин-ил)-метилН-имидазол (продукт В),4- (2 р 2 рб-триметилНр 3-бензодиоксин-ил)-метилН-имидазол (продукт Г)р4-1-(4 Нр 3-бензодиоксин-ил)этилН-имидазол (продукт Д),4- 1-(2 р 2-диметилНр 3-бензодиоксин-ил)"пентилН-имидазол (продукт Е);хлоргидрат 4- (2,6-диметокси-(1- 15метоксиэтил)-фенил)-метилН-имидазол (продукт Б)4- 1-(2 р 2-диметилНр 3-бензодионслн-В"ил)-этил)-1 Н-имипаэол (продукт 3),д-11-(4 Нр 3-бензодиоксин-ил) -этил -1 Н-имидаэол (продукт И). ГПротивоишемическая сердечная активностье251, У крысы, У анестезированной иторакотомизированной крысы коронарнаялигатура вызывает стабильную сердечную ишемию, которая выражается в увеличении зубца К эпикардической электрокардиограммы. Противоишемическоевоздействие продукта проявляется вуменьшении увеличения зубца К,Таблица 1 показывает для продуктов, подвергнутых испытанию, дозу(ЭД , мг/кг), которая, введенная 35внутривенно группе из 5 крыс, вызывает среднее уменьшение по крайней мерена 203 увеличения зубца К у всех животных. В качестве стандартных веществ используют следующие продуктыэ 40пропанолол 1-(изопропиламино)-3(1-нафтилокси)-2-пропанол,верапамил: Ь-/3- 2(-3,4-диметок-сифенил) -атил-и етиламино ) -пропил/3,4-диметокси-М-(1-метилэтил)-бенэолацетонитрил,нифедипин: диметиловый эфир 1,4 дигидро,6-диметил-(2-нитрофенил)3,5-пиридиндикарбоновой кислоты.2. У собаки. У живой собаки, носителе пневматической коронарной закупорки (осс 1 цдег)р коронарная окклюэиявызывает увеличение сегмента 8 Т эпикардической электрокардиограммы. Про"тивоишемическое воздействие соединения проявляется в уменьшении увеличения сегмента ЯТ.Таблица 2 показывает для продуктов,подвергнутых испытанию, дозу (ЭДор9. мг/кг), которая, введенная внутривенно группе из 10 животных, вызывает среднее уменьшение по крайней мере на 20 Х увеличения сегмента БТ у всех животных.Из этих табл. 1 и 2 следует, что полученные продукты обладают активностями, которые выше стандартных веществ. Токсичность.Токсичность предлагаемых продуктов определялась у самца мьцпи ПМР 1 с по" мощью теста Ирвина. Возрастающие дозы изучаемого продукта вводили интраперитонеально группам из трех мышей до тех пор, пока не была достигнута смертельная доза (доза, вызывающая гибель двух животных из трех в течение 48 ч).В таблице 3 дается смертельная доза, наблюдаемая для исследуемых соединений. Из табл. 3 следует, что эти соединения очень мало токсичны, причем смертельная доза намного выше активных дозФармацевтические композиции, включающие соединения, полученные согласно предлагаемому способу, могут быть введены орально, парентерально или ректально. Формула изобретенияСпособ получения замещенных 1 Н- нмидазолов общей формулы 1021 1 где К 4 р К , Ки Кз - одинаковые или разные, каждый водород или С-С - алкил, К - водород С-С-алкил или С-СА- алкок сил, один из радикалов У 1 и У 1 - водород, а дРУгой - 02)р 21 и 2 рр взЯтые по отдельности, обозначают оба Са-С- алкил, а вместе группа -СНар- или С(СН) -, или их солей присоединения нетоксичныхр фармацевтически приемлемых. кислот, о т л и ч а ю щ и й с я тем, что восстанавливают имидазольное соединение общей формулы 111662349 30 Таблица 2г ЭЛ , мг/кг Продукт 2 р 0,09 0,01 1где К, Кз и Й имеют указанные значения;К - водород или С 1-С- алкил;К. - радикал К 1, как указано, илитрифенилметил,К - радикал К, такой, как указано, или хлор, 15один из радикалов У и У - водород, а другой - радикал 02, причем 2 и 2 имеют указанные значения, и при необходимости, таким образом полученные 1 Н-имидазолы формулы Т пере водят в их соли присоединения нетоксичных, фармацевтически приемлемых кислот. 0,24 10 0,26 0,007 0,1 0,31 01008 0,53 Пропранолол Таблица 3 Продукт Смертельная доза,мг/кгТаблица 125ЭД , мг/кг Продукт 80 0,09 253 Б 0,25 0,24 0,26 0,03 (0,23 9., 4 7 О, 3 2 30 Г 258 Г 3 300 35 31,2 ПропранололВерайамил Нифедипин 1 70 Составитель Г.ЖуковаТехред М.Моргентал Корректор И.ЗРдейи Редактор Н.Яцола Заказ 2139 Тираж ПодписноеВНИИПИ Государственного комитета по изобретениям и открытиям при ГКНТ СССР113035, Москва, Ж, Рауаская наб., д. 4/5 Производственно-издательский комбинат "Патент"., г.ужгород, ул. Гагарина,101А. Получение бромпроизводных,:.предшественников металлоорганическихпроизводных.1. 8-Бром-б-хлорН,3-бензодиоксин.1 а. 2-Бром-хлорфенол. Этот продукт получается по известному способу,1 б. 8-Бром-б-хлорН,3-бензодиоксин. В смесь 229 мл концентрированной серной кислоты с 620 мл уксуснойокислоты, охлахщенную до 15 С, сразудобавляют 311 г параформальдегида(2,23 моль) 2-бром-хлорфенола, Поддерживают перемешивание в течение148 ч при 15 С. Реакционную средунейтрализуют 3, 1 л 7,8 н. водного раствора гидроксида натрия. Образующийся осадок отфильтровывают, отсасываюти затем растворяют в толуоле. Растворсушат над сульфатом натрия, затем перегоняют, Остаток перемешивают в гексане и оставляют кристаллизоваться,Получают 364 г 8-бром-б-хлорН,3 бензодиоксина. Выход 65,5 , т.пл.108-110 С.Вычислено С 38 47 Н 2 40.СВНВгС 10Найдено,: С 38,9, Н 2,51.2, 6-БромН,3 -бензодиоксин,Этот продукт получают как указано вп.1 б из следующих количеств реагентов; 3,8 л уксусной кислоты, 654 млконцентрированной серной кислоты,1 кг (5,78 моль) 4-бромфенола и 870 г(28,9 моль) параформальдегида. Продолжительность реакции 120 ч при 0 С.оПосле нейтрализации реакционной смеси раствором, содержащим 1350 г гидроксида натрия, растворенных в 13 лводы, образовавшийся осадок отфильт-ровывают, затем растворяют в 7 л толуола. Органическую Фазу сушат путемазеотропной перегонки, отфильтровывают горячей для удаления полимеровформальдегида и выпаривают при пониженном давлении. Остаток перегоняютпри пониженном давлении, Получают933 г 6-бромНЗ-бензодиоксина,оВыход 75 , т.кип. 80-90 С (О, 13 мбар),Продукт кристаллизуется путем перемешивания в смеси диизопропиловогоэФира с гексаном, т.пл. 43-47 С,ЯМР-спектр (СИС 1 З), й: 4,88 (2 Н,с, Аг-СН), 5, 24 (2 Н, с -ОСНО-),6,39 (1 Н д., 3 = 8,7 Гц, АгН), 7,1340 45 50 55 Зб. 8-Бром-б-хлор,2-диметилН,3-бензодиоксин, Сырой остаток (1490 г), выделенный в предыдущей стадии, растворяют в 20 л толуола и 4,68 л 2,2"диметоксипропана в присутствии 296 г монтмориллонита К 10 (свежедегидратированный путем азеотропной перегонки с толуолом). Температура смеси повьппается до 30 С, поддерживают перемешивание при этой температуре в течение 115 ч. Реакционную смесь фильтруют и удаляют толуол при пониженном давлении,Полученный остаток очищают перегонкой при пониженном давлении. Получают 527 г 8-бром-б-хлор,2-диметил Н,3-бензодиоксина 90 -ной чистоты (анализ путем тонкослойной хроматографии высокого давления). Т,кип.100-112 С (0,026 мбар). Общий выход 75 . (рассчитано по отношению к используемому 2-бром-хлорфенолу)(1 Н, м., АгН), 7,31 (1 Н, дд.,- 8,7 и 2,4 Гц, АгН) .3. 8-Бром-б-хлор,2-диметилН 5 1,3-бензодиоксинЗа. 3-Бром-хлор-оксибензолметанол. 880 г (4,241 моль) 2-бромхлорфенола растворяют в 8,2 л водного38 -ного раствора Формальдегида (около 100 моль), Охлаждают на ледянойбане, затем добавляют, порциями по100 г за один раз, 1380 г (24 моль)гидроксида калия. Добавление осуществляют за 150 мин при 18-23 С, Поддерживают еще перемешивание в течение2 ч при комнатной температуре, затемпостепенно нагревают на водяной банедо 40 С. Реакция слегка зкзотермичесокая и температура среды стаби.".изируется около 45 С. Продолжают перемешивание в течение 178 ч при 40 С.Реакционную смесь затем охлаждают,добавляют 2 л воды и подкисляют дорН 3 с помощью 1420 мл концентриро 25 ванной соляной кислоты, Экстрагируют2 л дихлорметана, затем еще шесть раз1 л дихлорметана. Органическую фазупромывают 2 л воды, сушат ее над сульфатом натрия и растворитель выпаривают при пониженном давлении.Сырой полученный остаток (1490 г)содержит около 68,83-бром-хлороксибенэолметанола и около 23,8 исходного непрореагировавшего 2-бром 354-хлорфенола. Эту смесь используют вследующей стадии.(1 Н, м., АгН), 7,4 (1 Н, м., АгН).4 6-Бром2-диметилН3-бен У У5зодиоксин4 а. 5-Бром-оксибензолметанол.Это соединение получают как указанов п.За из 4-бромфенола. После выпаривания дихлорметана, служащего дляэкстракции, получают 709 г остатка,который хроматографируют на 1,2 кгдиоксида кремния (элюирующее средство; дихлорметан). После выпариваниярастворителя получают 602 г остатка,15содержащего около 507 искомого 5-бром 2-оксибензолметанола (анализ путемтонкослойной хроматографии высокогодавления). Этот остаток используют вследующей стадии. 204 б. 6-Бром,2-диметилН,3-бензодиоксин,572 г полученного в предыдущей.стадии остатка растворяют в 12,5 лбезводного толуола и 2,94 л 2,2-диметоксипропана. Туда же добавляют186 г монтмориллонита К 10 и смесьперемешивают в течение 25 ч при ком"натной температуре, Отфильтровывают,органическую Фазу перегоняют при пониженном давлении и снова растворяютостаток в 600 мл толуола. Этот раствор пропускают через колонку, содержащую 3 кг оксида алюминия (элюирующее средство: 6 л толуола). Элюат выпаривают при пониженном давлении иостается 180 г остатка, который перегоняют при пониженном давлении. Получают 60 г 6-бром,2-диметилН,3 бензодиоксина. Т.кип. 110-130 С40(0,027 мбар), Выход, рассчитанный поотношению к используемому 4-брсмфенолу, составляет около 107.ЯИР-спектр (СОС 1), 8: 1,52 (6 Н,с С(СНз, 4,80 (2 Н, с., СН),6,71,(1 Н, д., 3 = 7,9 Гц, АгН), 7,077,5 (2 Н, м., АгН).5. 8-Бром,2,6-триметилН,3 бензодиоксин. Это соединение получают согласно известному способу. Еговыделяют в виде остатка, который используют в следующей стадии.6. 1-Бром,6-диметокси-З-метоксиметилбензол.ба. 3-Бром-окси-метоксибензоло 55метанол. В течение 75 мин при 0 С28 г (0,12 моль) метил-бром-окси 4-метоксибензоата растворенного в250 мл тетрагидрофурана, прикапывают 96к суспензии 6,06 г (0,159 моль) алюмогидрида лития в 100 мл тетрагидро"Фурана. Смесь перемешивают в течение3 ч при комнатной температуре. Затемдобавляют смесь 11,5 мл воды и 12 млтетрагидрофурана и подкисляют с помощью 28,6 мл концентрированной солянойкислоты, растворенной в 300 мл воды.Экстрагируют дихлорметаном. Органические Фазы промывают водой, сушат надсульфатом натрия и выпаривают их припониженном давлении. Остаток (22,9 г)используют в следующей стадии, Выход9 1 с 7ЯИР-спектр (СПС 1), Р: 3,88 (ЗН,с., ОСН), 4,72 (2 Н, с., СН), 6,45(1 Н, д., 3 = 8,6 Гц, АгН), 7, (1 Н,д., Л = 8,6 Гц, АгН).6 б. 2-Бром-метокси-метоксиме 1тилфенол. При комнатной температурев течение 90 мин перемешивали смесь22,8 г 3-бром-окси-метоксибензолметанола, 230 мл 2,2-диметоксипропанаи 23 г монтмориллонита К 10 (свежедегидратированный путем азеотропной перегонки с толуолом) в 200 мл толуола,После фильтрации и удаления растворителя, получают 24,1 г 2-бром-метокси-б-метоксиметилфенола, перекристаллизуемого из диизопропилового эфира.Выход 997., т.пл, 98-100 С,Вычислено, 7; С 43,72, Н 4,45,СН и ВгОНайдено, 7; С 44,32, Н 4,49.бв. 1-Бром,6-диметокси-З-метоксиметилбензол,24,1 г (0,098 моль) 2-бром-метокси-метоксиметилфенола и 27, гметилиодида растворяют в 200 мл ацетона, в присутствии 14,82 г(0,107 моль) карбоната калия. Этотраствор кипятят с обратным холодильником при перемешивании в течение2,5 ч. Охлаждают, отфильтровывают неорганические соли и ацетон, отгоняют..Остаток обрабатывают водой и экстрагируют дихлорметаном. ОрганическуюФазу сушат над сульфатом натрия и перегоняют ее при пониженном давлении.Получают 13,1 г 1-бром,6-диметокси 3-метоксиметилбензола. Выход 517.,т,кип, 92-115 ОС (0,005 мбар),ЯИР-спектр (СПС 13),8 : 3,4 (ЗН,с., ОСИ), 3,88 (ЗН, с ОСН), 3,916623477, 1-Бром,б-диметокси-(1-метоксиэтил)-бензол,7 а, 3-Бром-окси-метокси-р-метилбенэолметанол. В течение 80 мин ипри 13-18 С 64, 3 г (О, 262 моль) 3 - 5бром-окси-метоксифетофенона ввиде раствора в 560 мл тетрагидрофурана прикапывают к суспензии 13,89 г(0,367 моль боргидрида натрия в300 мл тетрагидрофурана. Затем продолжают перемешивать в течение 2 чпри комнатной температуре, Реакционную смесь разлагают добавлением 140 млтетрагидрофурана, содержащего 3 млводы, затем туда же добавляют еще60 мл воды, Среду подкисляют добавлением 415 мл водного 1 н. раствора соляной кислоты. Удаляют тетрагидрофуран при пониженном давлении и воднуюФазу экстрагируют четырехкратно дихлорметаном. Органические Фазы сушатнад сульфатом натрия и перегоняют.Остаток (70 г) используют в следующейстадии,7 б, 2-Бром-метокси-б-(1-метоксиэтил)-фенол. Это соединение получаюткак указано в п.бб из 65 г 3-бромокси-метоксиметилбензолметанола,полученного в предыдущей стадии. Полученный сырой продукт количественноиспользуют в следующей стадии,ЯИР-спектр (СРС 1 ), 3: 1,48 (ЗН,д.; Х = б,б Гц, СН), 3,35 (ЗН, с.,ОСН), 3,88 (ЗН, с., ОСН 3), 4,54 (1 Н,к , Т =. 6, 6 Гц, СН), 6,48 ( 1 Н, д., 35,Х = 8,6 Гц, АгН), 7,02 (1 Н, д , Х =8,6 Гц, АгН).7 в, 1-Бром,6-диметокси-З(1-ме- .токсиэтил)-бензол. Это соединение получают как указано в п.бв из 2-бромЗ-метокси-б-(1-метоксиэтил)фенола,выделенного из предыдущей стадии, Получают 56 г 1-бром,б-диметокси-З-.(1-метоксиэтил)бензола. Выход 77 ,т,кип. 95-120 С (0,001 мбар) . 45ЯП-спектр (СИС 1), Я = 1,42 (ЗН,д., Х = б,б Гц, СНз), 3,23 (ЗН, с.,ОСН 3), 3,88 (ЗН, с., ОСН.), 3,90 (ЗН,с., ОСН 3), 4,68 (1 Н, к , Х = б,б Гц,СН), 6 79 (1 Н, д, Т = 8,6 Гц, АгН), 507,38 (1 Н, д., Х = 8,6 Гц, АгН).Б, Получение 4-(Р-СО)-1 Н-мидазолов.1, 1-ТрифенилметилН-имидазолкарбоксальдегид. 551 а. 1-ТрифенилметилН-имидазолметанол. Это соединение получают поизвестному способу, Выход 71,3 ,т.пл. 236 оС 9 81 б. 1-ТрифенилметилН-имидазолкарбоксальдегид, Это соединение такжеполучают по известному способу с выходом 87,5 , т,пл, 190-198 С.2. 1-МетилН-имидазол-карбоксальдегид. Этот продукт получают поизвестному способу.3. 5-Метил-трифенилметилН-имидазол-карбоксальдегид,За. 5-МетилН-имидазол-метанол.Это соединение получают по известному способуВыход 59,3Зб. 5-Иетил-трифенилметилНимидазол-метанол. К раствору 100 г(0,673 моль) 5-метилН-имидазолметанола в 1,5 л диметилформамида при10-14 С, в течение 15 мин добавляют230 мл (1,659 моль) триэтиламина, Затем при 8-11 С вводят раствор, содержащий 192 г (0,69 моль) трифенилметилхлорида в 2 л диметилформамида.Реакционную смесь перемешивают втечение 2 ч, затем ее выливают на14 л льда. Продолжают перемешиваниев течение 1 ч, затем осадок отфильтровывают, промывают его с помощью блводы и отсасьвают. Этот осадок затемобрабатывают 4 л кипящего этанола инерастворившуюся фракцию отделяют путем горячей фильтрации, Таким образом получают первую порцию (19 г,т.пл. 255-260 С) целевого продукта.Спиртовый фильтрат фильтруют горячимна норите, затем концентрируют и охлаждают при перемешивании. Вторая порция целевого продукта. кристаллизуется. Ее отделяют отфйльтровьванием:28,3 г (т.пл, 255-262 С). Фильтратокончательно перегоняют при пониженном давлении и остаток растворяют в1 л смеси дихлорметана с метанолом:95:5 и полученный раствор очищаютпропусканием через колонну с 1,8 кгдиоксида кремния (0,2-0,5 мм) (элюирующее средство.: смесь дихлорметанас метанолом 80:20). Получают еще72, 1 г целевого продукта. В целом получают 119,4 г, содержащих практически один иэ двух изомеров возможныхположений 4 и 5, а именно 5-метил.трифенилметилН-имидазол-метанола.Выход 50,1Полученный продукт используют вследующей стадии.Зв. 5-Иетил-трифенилметилНимидазол-карбоксиальдегид. В течение 85 мин в присутствии 32,65 г(О, 0536 моль) продукта, полученного напредыдущей стадии, в 400 мл хлороформа. Соли марганца удаляют фильтрацией на дикалите и фильтрат перегоня 5ют. Остаток перекристаллизуют из200 мл этилацетата. Получают первуюпорцию 4,37 г целевого продукта. Вторую порцию 9,43 г получают еще послеконцентрирования меточных растворовдо объема 60 мл и кристаллизации. Выход 73,5 , т.пл. 195-196 С. Полученный продукт включает один изомер,Вычислено, %: С 81, 18, Н, 5,68,И 7,95., 15С г 4 Нго ИгОНайдено,: С, 81,37, Н 6,25,И 7,95.4. 1-(1-ТрифенилметилН-имидазол 4-ил)-1-этанон,204 а. 0 -Иетил-трифенилметилНимидазол-метанол. Это соединениеполучают по известному способу.46, 1-(1-ТрифенилметилН-имидаэол-ил)-1-этанон. 25В течение 90 мин кипятят с обратным холодильником смесь 221,3 г(2,544 моль) диоксида марганца и56,9 г (О, 161 моль) 0 ь-метил-трифенилметилН-имидазол-метанола, ра 30створенную в 2,5 л хлороформа. Затемораствор охлаждают до 50 С, отфильтровывают и хлороформ удаляют отгонкой.Остаток растворяют в 400 мл изопропилового спирта и раствор фильтруют горячим на норите.35Продукт кристаллизуется путем охлаждения. Получают 32,8 г 1-(1-трифенилметилН-имидазол-ил)-1-этанона,Выход 58 ., т.пл. 158-160 С.Вычислено,: С 81,82, Н 5,68,40И 7,95.С г 4 Нго ИгОНайдено,.: С 81,89, Н 5,65,И 790.45, 1-(1-ТрифенилметилН-имидазол 4-ил)-1-пентанон,5 а. ф,-н-Бутил-трифенилметилНимидазол-метанол,1В атмосфере аргона медленно растворяют 0,22 моль н-бутилмагнийбромида в 75 мл тетрагидрофурана добавляют к 67,6 г (0,2 моль) 1-трифенилметилП-имидазол-карбоксальдегида(получен как указано в п.16) в 500 млтетрагидрофурана. Температуру смесиподдерживают около 20 С путем охлажоения на ледяной бане. Когда добавление закончится, перемешивают еще 30 мин.при комнатной температуре, затем добавляют последовательно 11 гхлорида аммония и 100 мл воды, Экстрагируют дихлорметаном. ОрганическиеФазы сушат над сульфатом натрия, затем выпаривают при пониженном давлении, Остаток перекристаллизуют изсмеси этилацетата с диэтиловым эфиром(2:1). Получают 45,8 г К-н-бутилтрифенилметилН-имидазол-метанола,Выход 58 ., т.пл, 119- 120 С.Вычислено, %: С 81,82, Н 7,07,И 7,07,Сг 7 Нгв ИгОНайдено, %: С 79, 13, Н 6,82,И 6,65.56. 1-(1-ТрифенилметилН-имидазол-ил)-1-пентанон. Это соединениеполучают как указано в п,46 изб-нбутил-трифенилметилН-имидазолметанола, полученного в предыдущейстадии, Иаслянистый остаток, полученный после выпаривания хлороформа,очищают путем хроматографии на диоксиде кремния (элюирующее средство;дихлорметан), Получают 39 г 1-(1-трифенилметилН-имидазол-ил)-1-ентанона. Выход 85,3 , т.пл. 115-118 С.Вычисленор % С 82 у 23 у Н бр 60 уИ 7,11.Сг НгьИгОНайдено, %: С 82,18, Н 6,61,И 7,14,В. Реакция металлоорганическогопроизводного с 4-(К-СО)-1 Н-имидазолами,1. Ы -(6-ХлорН,3-бензодиоксин 8-ил)-1-трифенилметилН-имидазолметанол.В атмосфере азота 121 г (0,485 моль) 8-бром-б-хлорН,3-бензодиоксина, растворенного в 400 мл безводного тетрагидрофурана, прикапывают к суспензии 12, 16 г (0,5 моль) магния в 430 мл безводного тетрагидрофурана, доведенного до кипения.Когда добавление заканчивается,выдерживают еще при кипении с обратным холодильником в течение 0,5 ч, затем охлаждают до 40 С. Таким образом, об 0разовавшееся магнийорганическое соединение быстро добавляют к 164 г (0,485 моль) 1-трифенилметилН-имидазол-карбоксальдегида в виде раствора в 2 л тетрагидрофурана, предва-орительно нагретого до 40 С. Темпера. тура сама повышается в процессе добавления до 50 С. Продолжают переме 1662349 1235 шивание в течение 1 ч при 40 ОС. Затемореакционную смесь охлаждают до 0 Си разлагают ее добавлением 1 л насыщенного раствора хлорида аммония. Об"разовавшийся осадок отфильтровываюти промывают его метанолом и эфиром.Получают 140,7 г продукта, Этот продукт еще очищают путем перемешиванияв 1 л воды. Отфильтровывают и промы 10вают этанолом, затем диэтиловым эфиром. Получают 121,3 гь-(6-хлорН 1,3-бензодиоксин-ил)-1-трифенилметилН-имидазол-метанола. Выход49,2% т пл. 233-235 С.Вычислено, %; С 73, 15, Н 4,91,И 5,50,Сз 11 ебс 1 203Найдено, %: С 73,21, Н 4,94,Л 5,48.2. М,-(4 Н,3-Бензодиоксин-б-ип)- 2 С1-трифенилметилН-имидазол-метанол. Это соединение получают также,как и предыдущее, но из 6-бромН 1,3-бенэодиоксина и 1-трифенилметил 1 Н-имидазол-карбоксальдегида. Послеразложения реакционной среды продуктреакции экстрагируют дихлорметаном иперекристаллизуют его из изопропилового спирта. Путем хроматографии по"лученного после выпаривания маточных30растворов остатка рекуперируют ещевторую порцию продукта, Выход 53 ,т,пл. 165-167 оС.Вычислено, %: С 78,48, Н 5,48,И 5,91.С 31 Н 1 бИГОЗНайдено,: С 78, 19, Н 5,82,И 5,84.3. 0(,-(6-Хлор,2-диметилН,3 бензодиоксин-ил)-1-трифенилметил1 Н-имидазол-метанол.К суспензии 26,73 г (1 моль + 10%избытка) магния в 250 мл безводноготетрагидрофурана добавляют 2 мл дибромэтана и нагревают до 30 С для 45инициирования реакции. Затем прикапывают 277,5 л (1 моль) 8-бром-хлор 2,2-диметил"4 Н,3-бензодиоксина, растворенного в 250 мл тетрагидрофурана, так, чтобы температура среды не 50превышала 40 С. Добавление длитсяооколо 150 мин. Иагнийорганическоесоединение охлаждают до 10 С (частичное осаждение) и добавляют его к раствору 338 г (1 моль) 1-трнфеннлметилН-имидазол-карбоксальдегида в2,8 л тетрагидрофурана, предварительно охлажденного до 0 С. В процессеодобавления температура смеси постепенно повышается до 20 о С. Продолжаютперемешивание еще в течение 1 ч приэтой температуре, затем добавляют53,5 г (1 моль) хлорида аммония.Перемешивают в течение 1 ч, добавляют еще 18 мл воды и продолжают перемешивание дополнительно в течение1 ч. Удаляют тетрагидрофуран при пониженном давлении. Остаток обрабатывают 5 л дихлорметана и промывают2 л воды, содержащей 30 г бисульфитанатрия. Водную Фазу отделяют и промывают ее 1 л дихлорметана. Органические фазы промывают еще водой, затемсушат над сульфатом натрияи растворитель удаляют при пониженном давлении. Остаток перекристаллизуют примерно на 4 л толуола при 80 С и Фильтруют при нагревании на норите. Такицобразом получают 335, б г 6(, -(б-хлор,2 диметилН, 3-бензодиоксин-ил) -1 трЮенилметилН-имидазол-метанола,который содержит молекулу толуола.Т.пл. 120 С, затем 188 С.Вычислено,: С 6,81, Н 5,78,И 4,37.Су Нр С 1 ИуОз + С 7 Н 8Найдено, Ж: С 74,78, Н 5,24,И 4,73.Следующие соединения получают способом, описанным в п.З.4.0-(6-Хлор,2-диметилН,3 бензодиоксин-ил)-1-метилН-имида"зол-метанол. Из 8-бром-б-хлор,2 диметилН 1,3-бензодиоксина и 1-метил"1 Н-имидаэол-карбоксальдегида.Добавление магнийорганического соединения осуществляют при 0 С. Выходб63,53, т.пл. 131-136 С (этилацетат).Вычислено, %; С 58,35, Н 5,51,И 9,08, С 1 11,55.Ср Н 7 С 1 ИОНайдено,: С 58,47, Н 5,54,И 8,97, С 1 11,495. 0"(6-Хлор,2-диметилН,3 бензодиоксин-ил)-5-метил-трифенилметилН-имидазол-. 4-метанол. Соединение получают из 8-бром-хлор,2-диметилН,3-бензодиоксина и 5-метил.-трифенилметилН-имидазол-карбоксальдегида. Добавление магнийорганического соединения осуществляют при ОС. Выход 50 , т.пл. 100120 С (ацетонитрил),ЯМР-спектр (СРС 1 ) Р: 1,4 (6 Н, м.,СНЗ-С-СН), 1,94 (ЗН, с., СН), 4,796.О в (2,2,6-ТриметилН,3-бензодиоксин-ил)-1-трифенилметилНимидазол-метанол. Соединение получают иэ 8-бром,2,6-триметилН,3 бензодиоксина и 1-трифенилметилНимидазол-карбоксальдегида, Магнийорганическое соединение добавляют прикомнатной температуре, Продукт реакции очищают с помощью хроматографиина диоксиде кремния (15 тут) (Элюи,рующее средство: дихлорметан и метанол 98:2). Выход 31%, т.пл. 205215 С (ацетонитрил),ЯМР-спектр (СЭС 1), 2: 1,3 (ЗН,с., СН-С-СНэ), 1,38 (ЗН, с СН-ССН), 2,21 (ЗН, с., СНэ), 4,78 (2 Н,с., СН), 6,02 (1 Н, уширенный синглет, СНОН), 6, 72 (2 Н, м 1 тпН + ОН),7,0-7,65 (18 Н, м., АгН + 1 тпН).7. М -(2,2-ДиметилН,3-бенэоди 20оксин-ил)"1-трифенилметилН-имида;зол-метанол. Соединение получают из,б-бром,2-диметилН,3-бензодиоксина и 1-трифенилметилН-имидаэол 4-карбоксальдегида. Температура реакционной среды не превышает 40 С. Выход 54,5 Е, т.пл. 155-162 С (ацетонит.рил) .ЯИР-спектр (ДМСО), 8: 1,43 (6 Н,с СН-С-СНЭ), 4,77 (2 Н, с., СН),5,53 (2 Н, с., СН и ОН), 6,6-7,7 (20 Н,м., АгН + 1 шН).8.06-(2,6-Диметокси-З-метоксиметилфенил)-1-трифенилметилН-имидазол-метанол. Соединение получают из 351-бром,6-диметокси-З-метоксиметилбензола и 1-трифенилметилН-имидазол"карбоксальдегида. Продукт реакФции очищают с помощью хроматографиина диоксиде кремния и он находится в 40виде стекловидного лака. Выход 487.,ЯИР-спектр (СПС 1 э),3 : 3,36 (ЗН,с., ОСН), 3,7 (6 Н, с., 2 хОСН), 4,43(2 Н, с., СН), 6,0-6,4 (1 Н, м., СН),6,5-7,.6 (20 Й, м АгН + 1 шН + ОН), 459.0 - 2,6-Диметокси-З-(1-метоксиэтил)-фенил -1-трифенилметилН-имидазол-метанол. Соединение получаютиз 1-бром,6-диметокси-З-(1-метоксиэтил)бенэола и 1-трифенилметилНимидаэол-карбоксальдегида. Продуктреакции очищают хроматографией на диоксиде кремния (элюирующее средство;смесь дихлорметана с метанолом 98:2),Выход 49,7 Х, т.пл, 96-99 ОС (ацетонитрил),Вычислено, 7: С 76,4, Н, 6,36,Н 5,24.С МНм И 04 Найдено, Х: С 76,37, Н 6,31,М 5,29.10. Ы -(6-Хлор,2-диметилН,3 бензодиоксин-ил)-Мгметил-трифенилметилН-имидазол-.метанол,Иагнийорганическое соединение 8 бром-б-хлор,2-диметилН,3-бенэодиоксина получают как указано в п,З.Затем, за 20 мин и при температуре,не превышающей 25 С, добавляют 15 г(0,046 моль) этого магнийорганического соединения к 15 г (0,0426 моль)1-(1-трифенилметилН-имидазол-ил)-1-этанона в виде раствора в 150 млтетрагидрофурана. Перемешивают в течение 165 мин, затем разлагают реакционную среду с помощью 2,5 г хлорида аммония, растворенного в 50 мл воды. Экстрагируют дихлорметаном. Органическую фазу высушивают над сульфатом натрия, затем перегоняют. Остатокочищают с помощью хроматографии надиоксиде кремния (элюирующее средство: смесь дихлорметана с метанолом98:2), Получают 5,68 г 0-(6-хлор,2 диметилН,3-бенэодиоксин-ил)-О;метил-трифенилметилН-имидазолметанола. Т,пл, 182-184 С (этилацетат). Это соединение идентично полученному в примере 2 (Б,2).11.ф,-н-Бутил-Ы;(6-хлор,2-диметилН,3-бенэодиоксин-ил)-1-трифенилметилН-имидазол-метанол,Это соединение получают как и предыдущее соединение, вводят во взаимодействие магнийорганическое соединение 8-, бром-б-хлор,2-диметилН,3 бензодиоксина с 1-(1-трифенилметил 1 Н-имидазол-ил)-1-пентаноном. Выход54,6 Х, т.пл. 124 пС (петролейный эфир).ЯИР-спектр (СЭС 1), : 0,6-3,3 (15 Н,м., С 4 Н и СН-С-СЙэ), 4,26 (1 Н, уширенный с., ОН), 4,78 (2 Н, с., СН ),6,7-8,0 (19 Н, м., ЛгН + 1 тпН),12.М -(2,2-Диметил Н,3-бензоди-.оксин-ил)-1-метилН-имидаэол-метанол (хлоргидрат). Подвергают гидрогенолизу 15,88 гь.-(6-хлор,2-диметилН,3-бенэодиоксин-ил)-1-метилН-имидазол-метанола (полученв п.4), растворенного в 160 мл метанола, в присутствии 3 г 10%-ного палладия на угле, под давлением водорода3,5 бар при 50 С в течение 150 мин.Затем катализатор отфильтровывают,растворитель удаляют и остаток перемешивают в диэтиловом эфире. Эфирную5 10 15 20 25 30 35 40 45 50 55 П р и м е р 2. Получение исходного соединения формулы 11 (Кб - Н, К -алкил с С-С 4,илн трифенилметил).А. Получение кетонов;1, (6-ХлорН,3-бензодиоксин- .пл)-(1-трифенилметилН-имидазолил)-кетон,Это соединение получают согласноспособу, описанному в примере 1(Б4 б) изба-(6-хлорН-бензодиоксин 8-ил)-1-триЬенилметилН-имидазолметанола получен в примере 1,В,1).Вых.д 953 т.пл. 175-182 С. Образец,перекристаллизованный из этанола,плавится при 182-185 С и 203 С.Вычислено, %ф С 73,45, Н 4,54,Б 5,53, С 1 7,01.С 51 НС 1 БОЗНайдено, %: С 72 48, Н 4,48,М 5,18, С 1 6,96;2. (6-Хлор,2-диметилН,3-бензодиоксин-ил)-(1-трифенилметилНимидазол-ил)кетон. Это соединениеполучают по способу,описанномув примере 1,Б,4 б изб-(б-хлор-диметил 4 Н,3-бензодиоксин-ил)-1-трифенилметнлН-имидазол-метанола (получен в примере 1,В,З). Выход 88 . (продукт практически чистый), т,пл. 200205 С.Вычислено,.: С 74,08, Н 5,05,И 5,24, С 1 6,64.С, НС 1 Н,ОНайдено, %ф С 74,17, Н 5,03,И 5,22, С 1 6,73.Б. Реакция кетона с магнийорганическим соединением.1,р-(6-ХлорН-бензодиоксин 8-ил)метил-трифенилметилН-имидазол-метанол.Суспензию 0,148 моль метилмагнийиодида в 150 мл диэтилового эфира при30 д С, при комнатной температуре, добавляют к 17,3 г (0,033 моль) (6 хлорН,3-бензодиоксин-ил)-(1 трифенилметилН-имидазол-ил)кетона в виде раствора в 200 мл тетрагидрофурана. Температура реакционнойсреды повышается до 40 С. Когда до. бавление заканчивается, то продолжают перемешивание в течение 1 ч прикомнатной температуре. Добавляют 8 г, хлорида аммония и выдерживают еще приперемешивании в течение 1 ч. Послеэтого добавляют 100 мл воды и реакционную среду экстрагнруют два разадихлорметаном. Сушат органические фазы над сульфатом натрия и растворитель удаляют при пониженном давлении. Остаток перекристаллизуют из 100 млэтилацетата. Получают 13,77 г О-(бхлорН-бензодиоксин-ил)-о-метил-трифенилметил-.Н-имидазол-метанола. Выход 80% т.пл. 248-250 С(разложение)Вычислено,.: С 73,49, Н 5,15,И 5,36, С 1 6,7.СПСЩ,О,Найдено,: С 7341 Н 5,05,И 5,26, С 1 6,93.2.0 в (б-Хлор,2-днметилН,3 бензодиоксин-ил)метил-трифенилметилН-имидазол-метанол. Этосоединение получают, как и предыдущее соединение, из метилмагнийиодидаи (б-хлор,2-диметилН,3-бензодиоксин-ил)-(1-трифенилметилН-имидазол-ил) кетона. Полученный после выпаривания дихлорметана остаток кристаллизуется при перемешивании в диэтиловом эфире. Выход 80 , т.пл. 182184 С (этилацетат).Вычислено,.: С 4, 11, Н 5,63,.М 5,08, С 1 6,45.СНз С 110 зНайдено,.: С 73,98, Н 5,65,И 5,00, С 1 6,49П р и м е р 3. Получение исходного соединения формулы П (Кб. - Н илиалкил с С 1-С 4 КН)1. 4-(22-ДиметилН-бензодиоксин"8-ил)-(метокси)-метилсяН-имидазол-(хлоргидрат) и М-(22-диметил 4 Н,3-бензодиоксин"8-ил)-1 Н-имидаэол"метанол (хлоргидрат). Подвергают гидрогенолизу, 125,4 г (0,23 моль)О(-(б-хлор-диметилН-бензодиоксин-ил)-1-трифенилметилН-имида-эол-метанола (получен в примере1,В,З), частично растворенного в1250 мл метанола, в присутствии 6 г10%-ного палладия на угле, и под начальным давлением водорода 2,7 бар.Реакция осуществляется при 60 С.Затем катализатор отфильтровывают наЬуй 1 о-се 1 и метанол удаляют при пониженном давлении, Остаток обрабатывают 100 мл метанола и охлаждают на ледяной бане. Удаляют путем фильтрациитрифенилметан, который выкристаллизовывается и затем Фильтрат выпаривают. Полученный остаток перемешиваютв течение по крайней мере 12 ч в650 мл диэтилового эфира. Осадок отфильтровывают и промывают диэтиловымэфиром. Получают 64,59 г аморфногопорошка, который образован смесью целевого спирта и 0-метилированногопроизводного в соотношении 35/65, определенного по ЯИР. Эту смесь используют в следующей стадии. Образец5 36 г этой смеси продуктов перекрисФ5таллизуют из 20 мл смеси этанола сэфиром 1:1. Выделяют 1,35 г чистогохлоргидрата оЕ-(2,2-диметилН,3 бензодиоксин-ил)-1 Н-имидаэол-метанола. Этот продукт не имеет четкойточки плавления (разложения).Вычислено, %: С 56,66, Н 5,40,И 9,44, С 1 11,97.С 4 Н 16 И О НС 1Найдено, %: С 55,81, И 5,77,И 8,94, С 1 11.,83.0-Метилированное производное, которое получается в процессе гидрогенолиза, отделяют от реакционной смеси, нейтрализуют добавлением аммиакаи затем очищают с помощью хроматограФии, Полученный 4-(2,2-диметилН 1,3-бенэодиоксин-ил)-(метокси)-метипН-имидаэол превращается в хлоргидрат добавлением раствора соляной 25кислоты в метаноле. Т.пл. 150-155 Сф(получен в примере 1,В,5), растворенного в 300 мл метанола, в присутствии1,5 г 10%-ного палладия на угле, под 45давлением водорода 3,5 бар, при 50 С,.в течение 3 ч. Затем катализатор отфильтровывают, удаляют растворительи остаток перемешивают в диэтиловомэфире для удаления трифенилметана. 50Остаток, полученный после декантацииэфирнойфаэы,характеризуют ЯМР-спект-:ром, и он представляет собой смесь,включающую 65% хлоргидрата 0-метилированного производного и 35% хлоргидрата спирта. Эту смесь.используют вследующей стадии. .3. 4-(2,2,6-ТриметилН,3-бен-зодиоксин-ил) в (метокси)метилсяНимидазол и 4-(2,2,6-триметилН,3 бензодцоксин-ил)метилН-имидазол.Подвергают гндрогенолизу 9,45 г(0,0183 моль) Ы -(2,2,6-триметилН 1,3-бензодиоксин-ил)-1-трифенйлметилН-имидазол-метанола (полученв примере 1,В,6), растворенного в300 мл метанола, в присутствии 0,6 г10%-ного палладия на угле в течение4 ч при 80 С под давлением водорода2 бар. Затем катализатор отфильтровывают и растворитель удаляют, Полученный остаток перемешивают в диэтиловомэфире для удаления трифенилметана,затем хроматографируют на 700 г диоксида кремния (10 тут) (элюирующеесредство: дихлорметан:метанол 95;5).Выделяют смесь 40:60 4-(2,2,6-триметилН,3-бензодиоксин-ил)-метил 1 Н-имидазола и 4-(2,2,6-триметил 4 Н,3-бензодиоксин-ил)-(метокси)метил-Н-имидазола, идентифицированного с помощью ЯМР по наличию пика,соответствующего метокси-радикалу(ДМСО, о: 3,13)Эту смесь используютв следующей стадии.4. 0-(2,2-ДиметилН,3-бензодиоксин-б-ил)-1 Н-имидазол-метанол.Способ тот же, что и в п.2 но исходят иэ М-(2,2-диметилН,3-бензодиоксин-ил)-1-трифеннлметилН-имидаэол-метанола (получен в примере1,В,7). Полученный остаток хроматографируют на диоксиде кремния (15 тут)(элюирующее средство: дихлорметан-метанол 95:5). Выделенный 0-(2,2-диметилН,3-бензодиоксин-б-ил)-1 Нимидаэол-метанол перекристаллиэуютиэ этилацетата. Т.пл. 96 ОС.Вычислено, %: С 64,62, Н 6,15,Н 10,77.С 1411 6 ИОНайдено, %: С 64,37, Н 6,40,0 10,68,5. Ж - (2, 2-диметиЛН, 3-бенэодиоксин-ил)-Н-имидазол-метанол. Ксуспенэии 21,46 г (0,04 моль) О-(6 хлор,2-диметилН,3-бенэодиоксин 8-ил)-1-трифенилметилН-имидазолметанола (получен в примере 1,В,З) в2 л аммиака и 200 мл толуола, добав"ляют порциями 5,75 г (0,25 моль) нат-.рия. Выдерживают при перемешивании втечение 40 мин, затем реакционнуюсмесь разлагают добавлением 6,42 г
СмотретьЗаявка
4203647, 03.11.1987
ЮЦБ С. A
ЭРИК КОССМАН, ЖАН-ПЬЕР ГЕРТ, ЖАН ГОБЕР, ФИЛИПП МИШЕЛЬ
МПК / Метки
МПК: A61K 31/4164, A61P 9/10, C07D 233/58
Метки: 1н-имидазолов, замещенных, кислот, нетоксичных, приемлемых, присоединения, солей, фармацевтически
Опубликовано: 07.07.1991
Код ссылки
<a href="https://patents.su/15-1662349-sposob-polucheniya-zameshhennykh-1n-imidazolov-ili-ikh-solejj-prisoedineniya-netoksichnykh-farmacevticheski-priemlemykh-kislot.html" target="_blank" rel="follow" title="База патентов СССР">Способ получения замещенных 1н-имидазолов или их солей присоединения нетоксичных, фармацевтически приемлемых кислот</a>
(бензофуран-2-ил)-имидазолы, обладающие противогрибковой и антибактериальной активностью

Номер патента: 1600630
Опубликовано: 15.10.1990
Авторы: Адриано, Витторио, Данило, Карло, Марио, Россела
МПК: A61K 31/343, A61K 31/4178, A61P 31/10, C07D 405/06
Метки: активностью, антибактериальной, бензофуран-2-ил)-имидазолы, обладающие, противогрибковой
...модифицированный агар Сабуро при рН 7.Другие параметры не меняли,При использовании методики разведения в агаре приготавливали серийные разведения каждого из испытуемыхвеществ в дозе 128 - 0,03 мкг/мл в10 мл стерильного Физиологическогораствора, Затем по 2 мл из каждогоразведения брали и смешивали с 18 млстерильного модифицированного агараСабуро, находящегося в расплавленномсостоянии, а затем полученную Смесьвыливали в чашки Петри, К каждой серии с различными концентрациями испытуемого препарата прибавляли чашку с такой же средой, но без испытуемого вещества, в качестве контрольной, Каждую серию чашек каждого испытуемого вещества инокулировалимноготочечным инокулятором,Инокулят дерматофитов соскабливали, соблюдая асептику, с...
Способ получения фениламинометиленовых производных тионов пиразола или имидазола

Номер патента: 1384583
Опубликовано: 30.03.1988
Авторы: Алам, Квитко, Потапочкина
МПК: C07D 233/84
Метки: имидазола, пиразола, производных, тионов, фениламинометиленовых
...реакционной массы соляно-кислыманилином,П р и м е р 1. 33 г (0,15 М)Фенил-метил-Формил-хлорпиразола растворяют в 450 мл этанола в колбе, снабженной обратным холодильником, Добавляют к полученному раствору 50 мл (0,18 М) раствора Ба Б . Реакционную массу кипятят на водянойбане 1 ч, отгоняют этанол, остаток3растворяют в воде, профильтровыввютот нерастворившихся примесей, КФильтрату добавляют 39 г (0,30 М) соляно-кислого анилина Выдерживают,1 ч. Выпавший осадок отфильтровывают,промывают водой и сушат. Выход41,9 г (957,) т.пл. 150-153 С. Послекристаллизации из этанола т,пл.154155 С (т.пл. лит. 154-155 С),Примеры 1-10 различных условий4проведения реакции 5-хлор-альдегидапиразола с БаБ приведены в таблице.,П р и м е р 11. 308 г (0,15...
Способ получения производных бензиламина или их солей

Номер патента: 571188
Опубликовано: 30.08.1977
МПК: A61K 31/135, C07C 209/70, C07C 211/27, C07C 215/50, C07C 217/58
Метки: бензиламина, производных, солей
...и соединенные фильтраты выпаривают до 1 л.Эфирный слой экстрагируют (дважды) избыткомразбавленной соляной кислоты, а затем водой,Соединенные водные экстракты снова промываютэфиром, подщелоченным льдом и 50% - ным йаОН,и экстрагируют эфиром (дважды). Эфирные слоипромывают солевым раствором, сушат КгСОз ивьшаривают до образования масла (70 г), состоящего из двух основных компонентов. Масло (60 г)хроматографируют на колонке ЧЧое 1 пз (1,8 кг, нейт.ральной активности, тип 111), проявленной в бен.зол-гексане.(1:1). Бенэол-гексановой, бензольнойи ранней бензол-эфирной (10% - ный) фракциейэлюируют 34,2 г транс. продукта (свободное осно.ванне) с т.пл. 73 - 75 С.ЯМР (СОС 1 з): о 2,27 6 Н, синглет, й(СН,),3,05 1 Н, дублет, о = 3,5 - 4,0 сп.,...
Способ получения сульфамида

Номер патента: 342343
Опубликовано: 01.01.1972
Авторы: Иностранец, Ностранна, Соединенные
МПК: C07C 303/40, C07C 307/06
Метки: сульфамида
...15,4 г (0,11 моль) гексаметилентетрамина в хлороформе и перемешивают 24 час при комнатной температуре. Отфильтровывают выпавшую кристаллическую соль, фильтрат оставляют стоять на 24 час при комнатной температуре, выпавшие кристаллы присоединяют к ранее полученным. Соль, представляющую собой неочищенный 2,3,6-трихлорбензилгексаметиленбромид, вносят в 55 мл 6 и, соляной кислоты и перегоняют с водяным паром. Остаток в колбе подщелачивают 50%-ным едким натром и трижды обрабатывают серным эфиром. Эфирный экстракт сушат над сульфатом натрия, отгоняют эфир и перегоняют остаток в вакууме.2,3,6-Трихлорбензиламин перегоняется при 96 - 98 С/0,7 - 0,8 мм.4,2 г (0,02 моль) 2,3,6-трихлорбензиламина, 3,8 г (0,04 моль) сульфамида и 80 мл воды...
Способ получения производных соматостатина

Номер патента: 586837
Опубликовано: 30.12.1977
Авторы: Вернер, Ганс, Казимир, Манфред
МПК: A61K 38/31, C07K 14/655
Метки: производных, соматостатина
...с, 116-190.Приэритет по признакам;от 10.12. 73 - К -аминогрупа; Якарбэксильная гру(гна,от 01,08.74 - й. - водород, Я -во.дород, " -Н, Я -СОО; К - БН, В.=Нк Я Н й-СООН; Лиз ил Лиз --) -Лиз, Составитель В. ВолкомРедактор В, Мирзаджанова ТехредН,Андройчук Корректор.А. ЛакидаЗаказ 4652/716 Тираж 553 ПодписноеЦНИИПИ Государственного комитета Совета Министров СССРпо делам изобретений и открытий113035, Москва, Ж, Раушская наб., д. 4/5филиал ГПП ( Патент", г, Ужгород, ул. Проектная, 4зН-Трп Лиз(Бок)-Трн()-Фен-Трн()- -Сер()- К Н-СНЫ -СНе 8-,6 1 И), где Ыфимеет указанные значения; и- водород или группа Бут;3 /5 - группа Ацм или Трт,5и полученное линейное производное обшей формулыСНОСНЫ-СО-Гли-Цис(С )-Лиз(Бок)-...
Предыдущий патент: Способ получения призводных 2-оксоазетидина
Случайный патент: Акустооптический спектроанализатор