Вектор для клонирования генов, обеспечивающий регулируемую транскрипцию чужеродных генов, и способ его конструирования
Похожие патенты | МПК / Метки | Текст | Заявка | Код ссылки







Текст
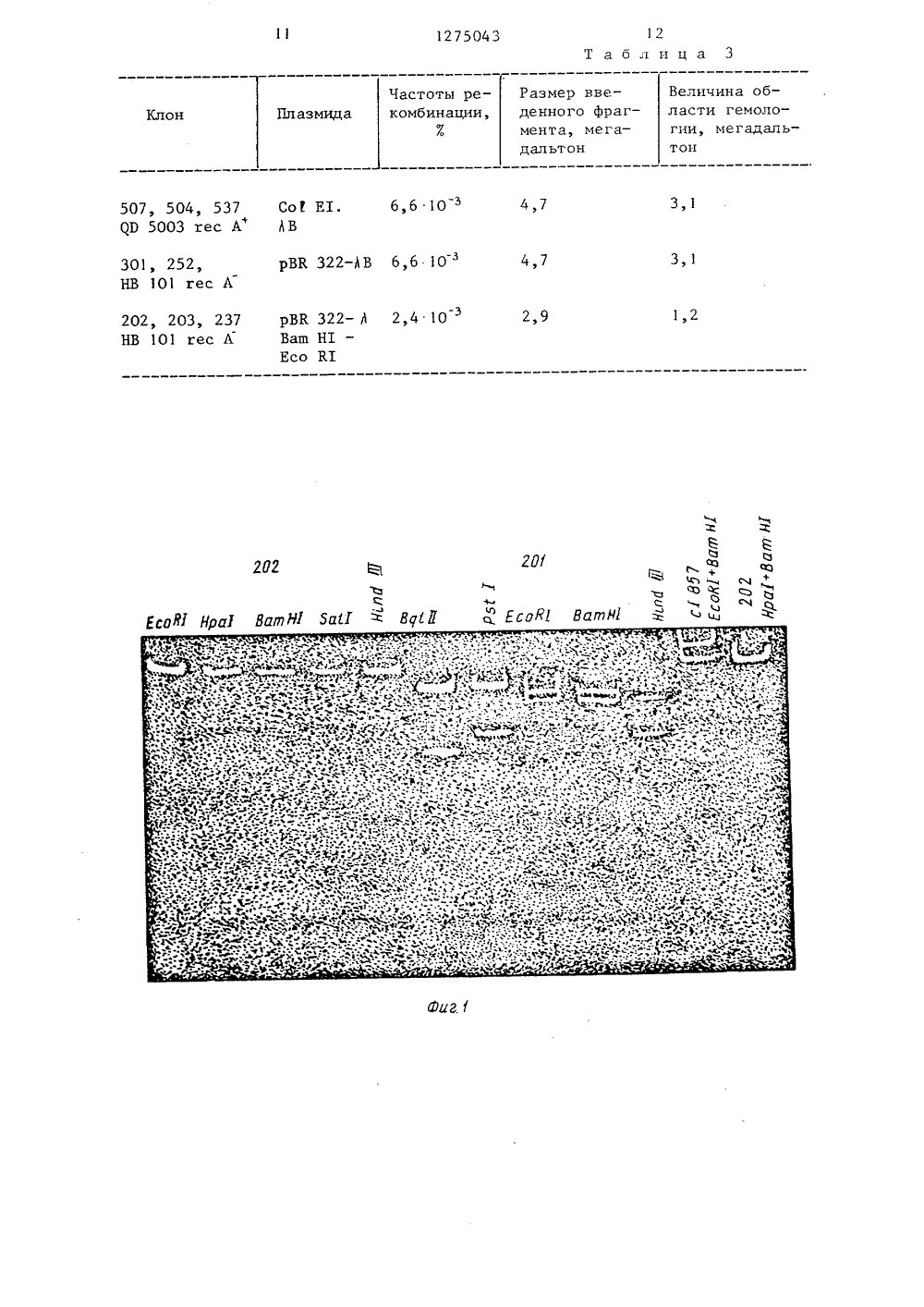
+ ОПИСАНИЕ ИЗОБРЕТЕНИЯК А ВТОРСКОМУ СВИДЕТЕЛЬСТВУ Цъ о ащ 6 ОСУДАРСТВЕННЫЙ КОМИТЕТ СССРПО ДЕЛАМ ИЗОБРЕТЕНИЙ И ОТКРЫТИЙ(71) Институт биохимии и физиологиимикроорганизмов АН СССР(54) ВЕКТОР ДЛЯ КЛОНИРОВАНИЯ ГЕНОВ,ОБЕСПЕЧИВА 10 ЩИЙ РЕГУЛИРУЕМУИ ТРАНСКРИПЦИ 10 ЧУЖЕРОДНЫХ ГЕНОВ, И СПОСОБЕГО КОНСТРУИРОВАНИЯ(57) 1. Вектор для клонирования генов, обеспечивающий регулируемуютранскрипцию чужеродных генов, СоСтоит из Ващ Н 1 - Есо К 1-фрагментаплазмидной ДНК рВК 322 и Ватп НдЕсо К 1-фрагмента ДНК регуляторной области фага , имеет мол. массу 5,3 ме -гадальтона, по одному сайту рестрикции эндонуклеаз рестрикции Ващ Н 1,Есо К 1, Бат 1, Нра 1, по два сайтаэндонуклеаз рестрикции Ряь 1 и Нтпй111, два промотора Р и Р , сайтыВащ Н 1 и Бат 1 удалены от Р -промотора на 0,7 и 0,9 мегадальтона соответственно, сайт Есо К 1 отстоит на0 7 мегадальтона от Р -промотора иУКна 3,1 мегадальтона от Р -промотора,ог - репликон плазмиды Со 1 Е 1, тер -мочувствительный репрессор с 1 857,гены фага й с 11, сго (атп 6), с 1,гех, 11, селективные маркеры Лна расстоянии 2 мегадальтона от Ващ Н 12сайта и АР на расстоянии 3,5 мегадальтона от Ватп Н 1-сайта, хозяин вектора - Еяс 1 тег 1 с 1 т 1 а со 1 т.2, Способ констртрования вектора для клонирования генов, включающий гидролиз донорной и реципиентной ДНК эндонуклеазами рестрикции, соединение образовавшихся фрагментов ДНК с помощью полинуклеотидлигазы, трансформацию клеток Еяс 1 тегт.с 1 тьа со 1 т. смесью полученных гибридных ДНК, отбор клонов, содержащих гибридные плазмиды, выделение вектора для клонирова - ния, о т л и ч а ю щ и й с я тем, что, с целью получения вектора с меньшей молекулярной массой, обеспечивающего регулируемую транскрипцию чужеродных генов, в качестве реципиентной ДНК используют ДНК плазмиды рВК 322, в качестве донорной ДНК - ДНК фага и с 1 857 сго 6, а гидролизу подвергают смесь донорной и реципиентной ДНК, из эндонуклеаз рестрикции используют эндонуклеазы Ващ Н 1 и Есо К 1, при этом последние вносят в реакционную смесь одновременно, отбор клонов, содержацих гидридные плазмиды, проводят на среде с ампициллином в присутствии фагов й с 1 и 1 т 434 ппщ,й с 1, причем из клеток выделяют вектор для клонироватвтя, содержащий ДНК регуляторной области фага ., включающей ранние промоторы фага 3 и гены репрессоров с мутациями с 1 857 и-35, Раушс роизводственно в полиграфическ прецприятие, г. Ужгород, ул. Проектная 6535/21 ВНИИПИ по 113035, Тираж Го суд ар лам изо осква,аг Ю 201 7 Ю 4 юмяу А ОЮ 1 АРГО гХ у О Г 5 Г Р Креня,ч Подписиитета СССРкрытийя наб., д, 4/1 12Изобретение относится к молекулярной биологии, а именно к векторудля клонирования генов, обеспечиваю -щему регулируемую, транскрипцию чужеродных генов, и способу его конструирования.Целью изобретения является получение вектора с небольшой молекуляр -ной массой, обеспечивающего регулируемую транскрипцию чужеродных генов.На фиг, 1 представлено электрофо -ретическое разделение Фрагментов ДНКпри гидролизе различными эндонуклеазами рестрикции; на Фиг. 2 - схемаплазмид 202 (с 1 сго ) и 203 (с 1 857сго 6) с точкой рестрикции и генетической картой; на фиг, 3 - кинете 2 )ка связывания репрессора с Р ДНК Фага й с 1 при различных температурах;на фиг, 4 - связывание репрессорас Р ДНК Фага т с 1 при различных тем -запературах; на Фиг, 5 - схема гибридных плазмид; на Фиг, 6 - кривая рос -та культур, содержащих различныеплазмиды при 30 С и при перенесениикультур на 43 С.Вектор для клонирования, обеспечивающий регулируемую транскрипциючужеродных генов, состоит из Ваш Н 1 Есо В 1-фрагмента плазмидной ДНК рВВ.322 и Ватп Н 1 - Есо Рс 1 в фрагмен ДНКрегуляторной области фага 3 и имеетмол. массу 5,3 мегадальтона, по одному сайту рестрикции эндонуклеазрестрикции Ватп Н 1, Есо К 1, БаР 1,Нра 1, по два сайта эндонуклеаз рест -рикции Рзс 1 и Нтпс 1 111, два промотора Р и Р , сайты Ватп Н 1 и Бат 1удалены от Р -промотора на 0,7 и0,9 мегадальтона соответственно, сайтЕсо тс 1 отстоит на 0,7 мегадальтон отР -промотора и на 3, мегадальтонаот Р -промотора, ого - репликон плазмиды Со 1 Е 1, термочувствительный репрессор с 1 857, гены фага Л с 11, сго(атп б), с 1, гех, Н, селективные маркеры и на расстоянии 2 мегадальто -на, от Ватп Н 1 - сайта и Ар на расстоя -нии 3,5 мегадальтона от Ватп Н 1-сайта,хозяин вектора - Езс 1 тегтс 1 тт.а со Рт.П р и м е р. Получение плазмиднойДНК р ВК 322,Клетки, содержащие плазмидурВР. 322, выращивают в 500 мл бу -льона до плотности О,б (510 клеток8на 1 мл), добавляют в культуральнуюсреду хлорамфеникол до конечной концентрации 200 мкг/1 мл и продолжают75043 1 О 15 20 25 30 35 40 45 50 55 культивирование в течение 12 ч. Затем клетки собирают центрифугирова - нием (5000 об/мин, 20 мин), промывают равным объемом буфера (50 мМ ттис -НСР, рН 8,0), суспендируют в 4 мл 20 в н сахарозы в 50 мМ теис.- НС 1, рН 8,0, добавляют 1,2 мл лизо - цима (5 мг/мл) и инкубируют 10 мин во льду при аккуратном помешивании, К смеси добавляют 24 мл 0,25 М ЭДТА рН 8,0, оставляют во льду на 1 О мин, а затем добавляют последовательно 2,6 мл 5 М НаС 1 и 1,2 мл 10%-ного додецилсутьфата натрия, инкубируют во льду в течение 5 - 7 ч. Осветленные лизаты получают центрифугированием при 16000 об/мин в течение 1 ч. К экстрактам добавляют СзС 1 из расчета 1,7 г/мл и бромид этидия до конечной концентрации 200 мкг/мл. Полученную смесь центрифугируют при 44000 об/ /мин в течение 24-48 ч, Нижнюю, прокрашенную бромистым этидием, зону собирают по каплям, прокалывая пробирку под зоной. Полученную ДНК диализуют против 10 мМ трис - НСР, рН 8,0мМ ЭДТА, 10 мМ НаСР.Получение ДНК Фага 1 . Культуру Е. соРт С 600 (Я с 1857 сго 6) выращи - вают до пгтотности 0,4 при 30 С, рез - ко прогревают на газовой горелке до 42 С и инкубируют при этой температуре в течение 15 мин, затем инкубируют при 37 С в течение 2 ч при интенсивной аэрации, добавляют 0,01 обь - ема хлороформа и через 10 мин лизаты освобождают от клеточных обломков центрифугированием. Фаг очищают тремя циклами дифференциального центрифугирования (20000 об/мин 3 ч, 6000 об/мин) и репротеинизируют водонасыщенным фенолом при рН 8,0 (50 мМ;Рис -НСР) дважды, Полученную ДНК фага 1 диализуют против 10 мМ тРис. - НСР, 1 мМ ЭДТА, 1 М НаСР трижды и окончательно тремя сменами 10 мМ трис -НСР, рН 8,0, 1 мМ ЭДТА при 20- кратном разведении.Рестрикция ДНК. Плазмидную (рВК 322) и Фаговую (1 с 1 857 сго 6) ДНК для рестрикции смешивают в соотношении 1:5 и одновременно добавляют эндонуклеазы рестрикции Есо К 1 и Вош Н 1 по 0,2 мкл на каждый мкг смешенной ДНК, Инкубацию проводят в 50 мМтгис НСР, рН 7,5, 10 мМ МССР , 1 мМ ЭДТА и 10 мМ дитиотреитоле (буфер А) в те. чение 1 ч при 37 С, а затем смесь3 1275 прогревают до 70 С в течение 10 мин. Анализ полноты гидролиза проводят с помощью электрофореза в 0,82 в н агарозе в тРис -боратном буфере, рН 8,3.Лигирование ДНК. Смешивают 225 мкл растворов фрагментов ДНК, полученных при гидролизе эндонуклеазами рестрикции (10 мкг), с 25 мкл 5 мМ АТФ и 2 мкл полинуклеотидлигазы (3000 ед/ /мл). Смесь инкубируют при 10 С в те-О чение 6 ч, при 10 в кратн разведении добавляют инкубационный буфер А, АТФ (конечная концентрация 0,5 мМ) и по - линуклеотидлигазу из расчета 1 мкл фермента на 10 мгк ДНК. Продолжают и 5 инкубацию при 0 С в течение 2 суток.Полученную смесь используют для трансформации.Трансформация рекомбинантной плазмидной ДНК бактериальных клеток. Ю 25 мл бульона заражают 0,1 мл ночной культуры Е. со 12. и выращивают до плотности 0,6. После 10 мин охлаждения клетки собирают центрифугированием и суспендируют в равном объеме 25 0,1 М МАЗО . Полученную суспензию охлаждают 20 мин, клетки собирают центрифугированием и суспендируют в 0,5 объема 0,1 М СаС 1 . Суспензию клеток охлаждают в течение 20 мин, 30 клетки осаждают центрифугированием и суспендируют в 1/50 исходного объема 0,1 М СаС 1 . После дополнительного2охлаждения (30 мин) клетки используют для трансформации. 35ДНК, предназначенную для трансформации, диализуют против 0,05 М СаС 1 с 10 мМ трис -НС 3, рН 7,5, смешивают с равным объемом СаС 1 -клеток и охлаждают в течение 20 мин, Затем смесь 40 подвергают 1 мин термической обработке при 42 С, выдерживают в течениео15 мин при 20 С и охлаждают в течение 20 мин. Эту смесь после десятикратного разбавления бульоном и под ращивания клеток при 30 С в течение 30 мин, наносят на чашки Петри с ампициллином (20 мкг/мл) и с Фагамии Л Ь 434 2.тпш Д (1 10 каждогомоФага на чашку). 50Анализ функционирования генов в гибридной плазмиде. Выросшие на твердой среде с ампициллином и в присутствии фагов колонии культивируют с 1 мл бульона в течение 5 ч при 30 С, 55 0,1 мл культуры высевают в мягком агаре на чашки с МПА, содержащим 20 мкг/мл ампициллина. На мягкий 043агар накапывают различные суспензии фагов от 1 10 до 1 10 фаговых час 5 6тиц в каплеРезультаты анализа представлены в табл. 1, знаком + отмечены фаги, способные размножаться на анализируемой культуре. Резистентность отобранных клонов к антибиотикам анализируют на агаризованной среде с антибиотиками ампициллином (20 мкг/мл) и тетрациклином (20 мкг/мл), При введении Фрагмента ДНК Фага в плазмнду между сайтами Есо 11 и Ваш Н 1 структурный ген, отвечающий за устойчивость клеток к тетрациклину, нарушается (табл. 1, резистентные к антибиотикам клоны отмечены знаком +). Отбирают пять клонов, резистентных к ампициллину, к фагус 1 и нечувствительные к г 11 мутантам фага Т 4.Выделение плазмид ускоренным методом, В связи с необходимостью одновременного анализа физической структуры плазмидных ДНК, выделенных из различных клонов, выделяют плазмиды ускоренным методом, который состоит в следующем.Культуру выращивают в 10 мл бульона, осаждают центрифугированием при 6000 об/мин в течение 20 мин, суспендируют в 1 мл 25 ь-ной сахарозы в 20 мМ тРис -НС 1 рН 8,0, добавляют 0,2 мл лизоцима (5 мг/мл) и 50 мкл ЭДТА (0,25 М). После инкубации при осторожном перемешивании в течение 10 мин во льду добавляют 50 мкл пан - креатической РНКазы (1 мг/мл), предварительно прогретой при 100 С в течение 5 мин, и 0,5 мл "лизирующей" смеси (0,3 мл 1 ОЕ тритон-Х, 7,5 мл М ЭДТА рН 8,0, 1,5 мл 1 М трис - НСЕ рН 8,0, 0,7 мл Н О). После инкубации2при постоянном помешивании стеклянной палочкой во льду в течение 10 мин получают осветленный лизат центрифугированием пробы в течение 1 ч при 17000 об/мин (КапегзЫ 2.). Полученный лизат разбавляют водой вдвое, обрабатывают равным объемом фенола, насыщенного 50 мМ тРис -НС 1, рН 8,0, при комнатной температуре в течение 10 мин, водную фазу собирают центриФугированием при 6000 об/мин. Остав - шийся в пробе Фенол удаляют эфирной экстракцией, а ДНК переосаждают 707. - ным спиртом.5 1275Рестрикционный анализ рекомбинатных плазмидных ДНК. Для изучения физической структуры ДНК и идентификации введенного фрагмента регулятор -ной области Фага 1 плазмидную ДНК обрабатывают эндонуклеазами рестрикцииЕсо К 1 Ватп Н 1, Ба 1 1, РзС. 1 Н 1 пй111, РЬа 1 и ВдГ 11,К 0,5 мкг ДНК в буфере, содержащем 50 мМ трис -НС 1, рН 7,5, 10 мМ 1 ОМцСГ, 10 мИ ДТТ, 1 мМ ЭДТА, добавляют 0,1-5 мкл эндонуклеазы рестрикции.Общий объем реакционной смеси колеблется между 10 и 40 мкл при обработ -ках различными рестриктазами. При 15совместном гидролизе оба Фермента добавляют к реакционной смеси и инкубируют одновременно. Продопжительностьинкубации изменяется в зависимостиот активности эндонуклеаз от 30 мин 20до 3 ч. Для всех эндонуклеаз рестрикции инкубацию проводят при 37 С, кро -ме гидролиза плазмидной ДНК Рв 1,который ведут при 30 С. Гидролизатыанализируют с помощью электрофореза 25реакционной смеси в 0,8 Е-ном агарозном пластинчатом геле в теис -боратном буфере рН 8,3.Результаты рестрикционного анализа представлены на фиг, 1 и. 2. 30Основываясь на известном расположении промоторов фага и данных рест -рикционно го анализ а, можно з аклю -чить, что сайты Ватп Н 1 и Ба 1 1 удалены от Р -промотора на 0,7 и 0,9 мегадальтона соответственно. Сайт эндонуклеазы рестрикции Есо Р 1 отстоит на 0,7 и 3,1 мегадальтона соответственно от Р и Р . Эти расстоянияи ьможно считать достаточно небольшими, 40так как транскрипция, начинаясь в области иммунности, продолжается влеводо поздних генов фага (ЗОБ фаговойДНК или около 10 мегадальтон) и вправо до гена 0 (около 5 мегадальтон), 45Анализ синтеза репрессора в клетках, содержащих плазмиды. Количестворепрессора в клеточных экстрактах оп -ределяют по методу Эхолса и Грин, которьп основан на анализе количества 50меченой фаговой ДНК, связываемой срепрессором и не задерживаемой нанитроцеллюлозных фильтрах, Этот метод использован для изучения кинетики связывания репрессора, выделенного из различных клонов, а также дляизучения уровня его связывания с ДНКпри различных температурах. 043 аНа Фиг. 3 показано, что кинетикасвязывания репрессора, выделенногоиз клеток с гибридными плазмидами,содержащими с 1 дикого типа, сущест -венно не зависит от температуры, тогда как связывание термочувствительного репрессора, синтезированного вклетках с плазмидами, несущими му -тантную регуляторную область фага,заметно ухудшается при незначительном изменении температуры инкубации,Уровень связывания репрессора при22 С не зависит от наличия мутациии принят за 1003.На фиг. 4 показано, что степеньсвязывания термочувствительного репрессора с ДНК быстрее уменьшаетсяпри увеличении температуры, чем репрессора дикого типа.Генетический анализ регуляторнойобласти фага введенной в рекомбинантную плазмидную ДНК, Для доказательства одновременного присутствиямутаций в генах с 1 и сго проводят ре.комбинацию дп чло между плазмидамии фагом 1 1. 434 с, который дает набактериальном газоне бляшки с мутнымцентром за счет эффективной лизогенизации. Штаммы, содержащие плазмидную ДНК, выращивают в 10 мл бульонадо плотности 0,2 (1,6 10 клеток/мл)8при 30 С, инфицируют фагом И. 434 смножественностью 0,1, инкубируют при30 С в течение 4 ч, добавляют 1/100объема хлороформа и оттитровывают набактериальном газоне, содержащем ине содержащем профаг 434. Наличие рекомбинантов дополнительно указываетна присутствие регуляторной областифага с 1 в гибридных плазмидах. Час -тоту рекомбинации анализируют сравнением титра Фагов на этих двух культурах при 37 С,Результаты опытов сведены в табл,З.Частоты рекомбинации соответству -ют теоретически рассчитанному значению и строго зависят от величины введенного в плазмиду фагового фрагмен -та. Кроме того, показано, что часто -та рекомбинации не зависит от генагес А Е. со 1, Гибридный фаг, полу -ченный при рекомбинации с плазмидамиклонов 203, 237, 252, образует на газоне клеток Е, соЕ С 600 при 37 Сопрозрачные бляшки, а при 30 - мутные, что указывает на наличие термочувствительной мутации в гене с 1,тогда как рекомбинанты с плазмидами7 12750 202, 301 и 537 при 37 С образуют мутные бляшки. Анализ наличия в гибридных фагах супрессируемой мутации в гене сго проводят сравнением эффективности титрования этих фагов на штаммах Е. со 2 с различными супрессорными т-РНК при 30, 38 и 43 С.Результаты анализа, сведенные в табл. 2, указывают на одновременное присутствие мутаций в генах сго и с 1, 1 О а также на различную степень супрес - сии в зависимости от типа присутствующего в штамме супрессора. По данным этой таблицы можно заключить, что гибрьщная плазмида содержит фрагмент 15 ДНК фага П с генами с 1 857, сго 6.Доказательство наличия термоиндуцированной транскрипции с левого фагового промотора в плазмиде, содержащей регуляторную область фага. При отО боре клонов, содержащих гибридные плазмиды, наряду с плазмидами типа 202, 203, 237, отбирают плазмиды с мол. массой 7,3 мегадальтон, описанные в данном примере 301 и 252 25 (фиг. 5). Рестрикционным анализом установлено, что эти гибридные плазмиды содержат Есо К 1 - фрагмент фагас регуляторными генами. Кроме описанных генов этой области плазмиды Зо содержат гены с 111, 1 сд 1, 1,и Еа 10. Обнаружено, что клоны, содержащие эти плазмьщы, очень плохо растут прио37 С. Изучают эффективность титрования этих клонов при различных температурах. Титр клеток при выращиваонии этих культур при 43 С понижается приблизительно на четыре порядка (фиг. 6 В) . При сравнении кривых роста штаммов (фиг, 6), содержащих раз личные гибридные плазмиды, показано, что эффект понижения титра клеток заВисит от наличия дополнительных генов и присутствия термочувствительной мутации в гене с 1, которая позволяет дерепрессировать транскрипцию с левого фагового промотора (Р ). Подобный эффект описан для лизогенных культур с 600 ( А с 1 875 атпР), и картиро - вано местоположение гена, отвечающе- о го за убийство клеток. Этот ген рас - положен в Н - опероне между с 111 и назван 11. Присутствие этого гена в плазмидах оказывает аналогичное дей - ствие. При непермиссивной температу ре наблюдается накопление сильно удлиненных клеток и отсутствие прямого лизиса. Отмечено сохранение характе -43 8ра кривой роста для клонов, переживших повьппенную температуру в течение6 ч. Полученные данные указывают наналичие в Есо К 1 в фрагмен функцио -нирующего гена 11.1, подчеркивают егонегативное влияние на рост клетокпри 37-42 С, содержащих соответствующие плазмиды, и доказывают зависимость интенсивности транскрипции сфагового промотора Р от температу -ры для генов, расположенных слева отэтого промотора,Таким образом, предлагаемый спо -соб конструирования плазмиды позволяет получить мультикопийную плазми -ду, содержащую сильные промоторы срегулируемой транскрипцией.Небольшую молекулярную массу, наличие четырех уникальных сайтов (ВашН 1, Ба 1 1, Есо К 1, Нра 1) рестрикциии их малая удаленность от соответствующих фаговых промоторов позволяетшироко использовать эту плазыщу дляклонирования чужеродных генов. Необходимо отметить присутствие близкорасположенных сайтов эндонуклеаз рестрикции Ваш Н 1 и Ба 1 1, которые позволяют клонировать фрагменты ДНК, пропродуцируемые такими эндонуклеазами,как Ваш Н 1, Вд 1 11, ВсГ 1, МЬо 1,Яа 1 (ХЬо) за счет идентичности "лип -ких" концов при различной специфичности узнавания, а также фрагменты,получаемые при совместном гидролизе соответствующей парой эндонуклеаз. При использовании пары Хйо 1Вд 1 11 или ХЬо 1 - ВсГ 1 наблюдается исчезновение сайтов рестрикции,что позволяет проводить соединениефрагментов в присутствии эндонуклеаз,не удаляя мелкий фрагмент (фиг.1). Полученные синтетические "линкеры" обеспечивают возможность клонирования также с помощью других эндонуклеаз рестрикции,Использование репликона мультикопийной плазмиды значительно увеличивает дозу клонируемых генов, вприсутствии мутаций в гене с 1 и сгообеспечивает возможность регуляциитранскрипции в широких пределах. Вприсутствии хлорамфеникола количество копий плазмиды в клетке можетувеличиваться на два порядка и теоретически достигать 452 тотальнойклеточной ДНК,Присутствие в плазмице гена 11,продукт которого является антитер9 12 минатором, обеспечивает в ряде случаев интенсивную транскрипцию на значительном протяжении, даже когда клонируемый фрагмент ДНК содержит определенные терминирующие кодоны, По приблизительной оценке подобные векторы позволяют увеличить синтез белкового продукта в сотни раз.Наличие регулируемой транскрипции позволяет при необходимости клонировать гены, вызывающие гибель бак - териальных клеток, и получать значительные количества продуктов этих генов при повышении температуры.Известны примеры, когда введение эукариотных генов в векторную ДНК Т аблица фаги7434 Ь 434 Й Х 1.21 Лтпг Тг 11 Т 4 Ар Те Рекомбинатныеплазмиды,0 1,0 1,0 1,0 1,0 0,47 0,03 О,5 А П. 203 х Ь 0,052 ,434 0,01 0,08 0,05 0,1 0,4 0,01 0,03 0,1 0,07 с 1 857 сго 6 0,04 0,3 П р и м е ч а н и е, В нижней части табл, 2 представлены известные данные. 201 с 1 202 с 1 203 с 1 252 с 1 301 сТ 301 с 1 304 сТ 75043 10фага 1 в отличие от тех же генов всоставе плазмиды дает транскрипциюэтих генов под контролем левого промотора фага , Это позволяет пред -положить, что описанная в примереплазмида должна обеспечить эффективную транскрипцию, по крайней мере,некоторых генов эукариотного происхождения.10 Предлагаемая плазмида является специализированным вектором, с помощью которого возможно создание штаммовсуперпродуцентов различных белков, а также некоторых других веществ био - логического происхождения.12 1275043 Величина области гемолоПлазмида Клон гии, мегадальтон 6,6 10507, 504, 537 ЯЭ 5003 гес А СоГ Е 1,ЬВ 4,7 3,1 рВК 322-АВ 6,6 10301, 252,НВ 101 гес Л 3 з 1 2,4 10202, 203, 237НВ 101 гес А 2,9 1,2 гаг Э ь Есор Нрав ВащМ 5 ай 1Вдй Р ВатН 1 4 ст, Есай 1 рВК 322- ЯВата Н 1 -Есо К 1 Частоты рекомбинации,7. Т аб тица 3 Размер введенного фрагмента, мегадальтон
СмотретьЗаявка
2664904, 13.09.1978
ИНСТИТУТ БИОХИМИИ И ФИЗИОЛОГИИ МИКРООРГАНИЗМОВ АН СССР
СОЛОНИН АЛЕКСАНДР СЕРГЕЕВИЧ, ТАНЯШИН ВАЛЕРИЙ ИВАНОВИЧ, СЕМЕНОВА ЛИДИЯ МИХАЙЛОВНА, БАЕВ АЛЕКСАНДР АЛЕКСАНДРОВИЧ
МПК / Метки
МПК: C12N 15/00
Метки: вектор, генов, клонирования, конструирования, обеспечивающий, регулируемую, транскрипцию, чужеродных
Опубликовано: 07.12.1986
Код ссылки
<a href="https://patents.su/10-1275043-vektor-dlya-klonirovaniya-genov-obespechivayushhijj-reguliruemuyu-transkripciyu-chuzherodnykh-genov-i-sposob-ego-konstruirovaniya.html" target="_blank" rel="follow" title="База патентов СССР">Вектор для клонирования генов, обеспечивающий регулируемую транскрипцию чужеродных генов, и способ его конструирования</a>
Рекомбинантная плазмидная днк ртnf 22, содержащая полусинтетический ген фактора некроза опухоли человека промежуточная плазмида для конструирования рекомбинантной плазмидной днк ptnf 31, кодирующая полипептид с
Номер патента: 1438240
Опубликовано: 20.03.1996
Авторы: Болдырева, Добрынин, Коробко, Кравченко, Недоспасов, Турецкая, Филиппов, Чувпило, Шахов, Шингарова
МПК: C12N 1/21, C12N 15/28
Метки: ген, днк, кодирующая, конструирования, некроза, опухоли, плазмида, плазмидная, плазмидной, полипептид, полусинтетический, промежуточная, рекомбинантная, рекомбинантной, ртnf, содержащая, фактора, человека
1. Рекомбинантная плазмидная ДНК pTNF 22, содержащая полусинтетический ген фактора некроза опухоли человека, - промежуточная плазмида для конструирования рекомбинантной плазмидной ДНК pTNF 31, с мол.м. 3,6 MD (5,95 т.п.о. ), содержащая:Bam I - Hind III-фрагмент ДНК плазмидного вектора PVR 291 с промотором, оператором и измененным геном -галактозидазы, кодирующим 1027 аминокислотных остатков размером 5,35 т.п.о.;Bam I - Msp I-фрагмент синтетической ДНК с участком, инициации трансляции Escherichia coli, инициирующим кодоном ATG и 31 N-концевым кодоном фактора некроза опухоли размером 120 тыс. п.о.;Msp I - Hind III-фрагмент ДНК рекомбинантного фага l 422 с...
Штамм еsснеriснiа coli в834 (, ), несущий плазмиду rsf 2124, -тест субстрат для рестрикционной эндонуклеазы ecor11

Номер патента: 908793
Опубликовано: 28.02.1982
МПК: C12N 1/02
Метки: 2124, ecor11, в834, еsснеriснiа, несущий, плазмиду, рестрикционной, субстрат, тест, штамм, эндонуклеазы
...так в органической форме в виде пептона, аминокислот, Нитраты восстанавливает донитритов.40Желантину не разжижает.Урсазная активность не обнаруживается.Индол не образует,Проявляет устойчивость к антибиотикам -ампициллину (25 мкг/мл).П р и м е р 1. Выледение плазмидной45ДНК,Клетки, содержащие плазмиду, выращиваютв 500 мл бульона до плотности 0,6 (ФЭК,светофильтр Мф 6, кювета 0,5 см). Затемклетки собирают центрифугированием (600020 мин) суспещируют в 4 мл 25%-нойсахарозы в 50 мм трис-НС 1, РН 3,0,добавляюз 1,2 мл лизоцима (1 О мг/мл) иинкубируют И мин во льду при перемешивании,К смеси добавляют 2,4 мл 0.25 М ЭЛТА55(р Н 8,01, оставляют во льду на 10 мин, аэмем попзяляюг последовательно 2,6 мл 5 МРастдора ЧаС и 1,2 чл А-ного расгвора...
Способ конструирования плазмидной днк, штамм -продуцент эндонуклеазы рестрикции и способ получения эндонуклеазы рестрикции

Номер патента: 1040791
Опубликовано: 07.02.1985
Авторы: Баев, Глатман, Кравец, Кузьмин, Мороз, Солонин, Таняшин
МПК: C12N 15/00
Метки: днк, конструирования, плазмидной, продуцент, рестрикции, штамм, эндонуклеазы
...Физической структурыплазмидную ДНК выделяют ускореннымметодом (К 1 е 1 п Р,1 ейа 1, 1980,Р 1 азтпЫ, 8, 88-93) .Рестрикционный анализ проводят спомощью эндонуклеаз Р-1, Вя 1 11,11, Отобранные. клоны проверяютна присутствие системы рестрикциимодификации Есо 87. Для этого сравнивают эффективность посева (э.п.)фага; 0 на клетках, содержащихрекомбинантные плазмиды, а такжештаммах С 5183 (гь. -где ) и ) С 5183( г т+ ) с плазмидой рЬ 13. Наличие в клетках модификации устанавливают при снижении э.п. методом(Т. Ага, А.Ао 1 с 1, 3.Васй. 5, 18,1962) . Полученные данные представлены в табл. 2,Как видно из табл. 2, штамм3 С 5183/С 113 на четыре порядка ограничивает размножение фага Ъ0по сравнению со штаммом 1 С 5183( ГО ), не несущем плазмидуу...
Рекомбинантная плазмидная днк рук11, кодирующая рестриктазу и метилазу е corii, способ ее конструирования и штамм бактерий еsснеriснiа coli продуцент рестриктазы и метилазы е corii

Номер патента: 1453896
Опубликовано: 30.09.1990
Авторы: Бурьянов, Витенене, Косых
МПК: C12N 15/00
Метки: corii, бактерий, днк, еsснеriснiа, кодирующая, конструирования, метилазу, метилазы, плазмидная, продуцент, рекомбинантная, рестриктазу, рестриктазы, рук11, штамм
...штаммаЕ, соН В 834(рС 857),обусловлен тем,что штамм не содержит интерферируюих с ферментами Есо КП примесей исодержит плаэмиду с геном термочувст Овительного репрессора с 1 фага ляйбда,Содержаний рестриктаэы и метилазцЕсо К 11 в сконструированном штаммеравно 500000 ед, каждого фермента наг сырой биомассы клеток,Штами ЕвсИехсЫа со 1. ВКИ СК 2889 характеризуется следующими признаками;Иорфологические признаки.6 6ную как р 7 К 11 и содержащую в своем . составе трк фрагмента, которые обра,зуются при гидролизе плазмидной ДНК эндокуклеаэой рестрикции Рве 1Выделенную .ДНК р 7 К 11 трансформируют в штамм Е. со 11 В 834/рС 1857 Трансформаиты отбирают на среде, содержащей ампицкплин (50 мкг/мл) и канамкцин (25 мкг/мл). Плаэмида РС 1857 несет...
Способ конструирования рекомбинантной плазмидной днк, кодирующей фермент деацетоксицефалоспорин с синтетазудеацетилцефалоспорин с синтетазу

Номер патента: 1739856
Опубликовано: 07.06.1992
Авторы: Поль, Стефен, Сьюллен, Томас
МПК: C12N 15/52
Метки: деацетоксицефалоспорин, днк, кодирующей, конструирования, плазмидной, рекомбинантной, синтетазу, синтетазудеацетилцефалоспорин, фермент
...связуйщей 5 молекулы обрабатывают Т 4 ДНК"кинаэойи затем аннелируют с.образованиемнеповрежденной связуюцей молекулы.1 мкл гидролизованной Ваш Н 1 ДНКплазмиды рМЬС 12 лигируют с 4 мкл 1 О Бсо 1 - Нсо 1 фрагмента плазмидырЬС 1 и 10 мкл аннелированной связующей молекулы.Лигазной смесью трансформируютЕ.со 1 Аликвоты трансформированной 15 клеточной смеси вжевают на чашкахс Ь-агаром, содержащим 25 мкг/млхлорамфеникола, 40 мкг/мл Х-да 1.и40 хмкг/мл 1 РТС. Чашки инкубируютнри 37 С в течение ночи. Колонии,окоторые содержат плазмиду беэ вставки, такие как Е.со 1 К 12 М 109//рМЬС 12, проявляют синий цвет наэтих чашках. Колонии, которые содержат плазмиду с вставкой, такие какЕ.со 11 К 12 1 М 109/рРБ 53, проявляютбелый цвет на этих чашках,...
Предыдущий патент: Способ производства сухих питательных сред
Следующий патент: Способ автоматического управления процессом выращивания микроорганизмов
Случайный патент: Устройство для центробежного формования изделий