Способ получения серусодержащих органических соединений или их солей
Похожие патенты | МПК / Метки | Текст | Заявка | Код ссылки


















Текст
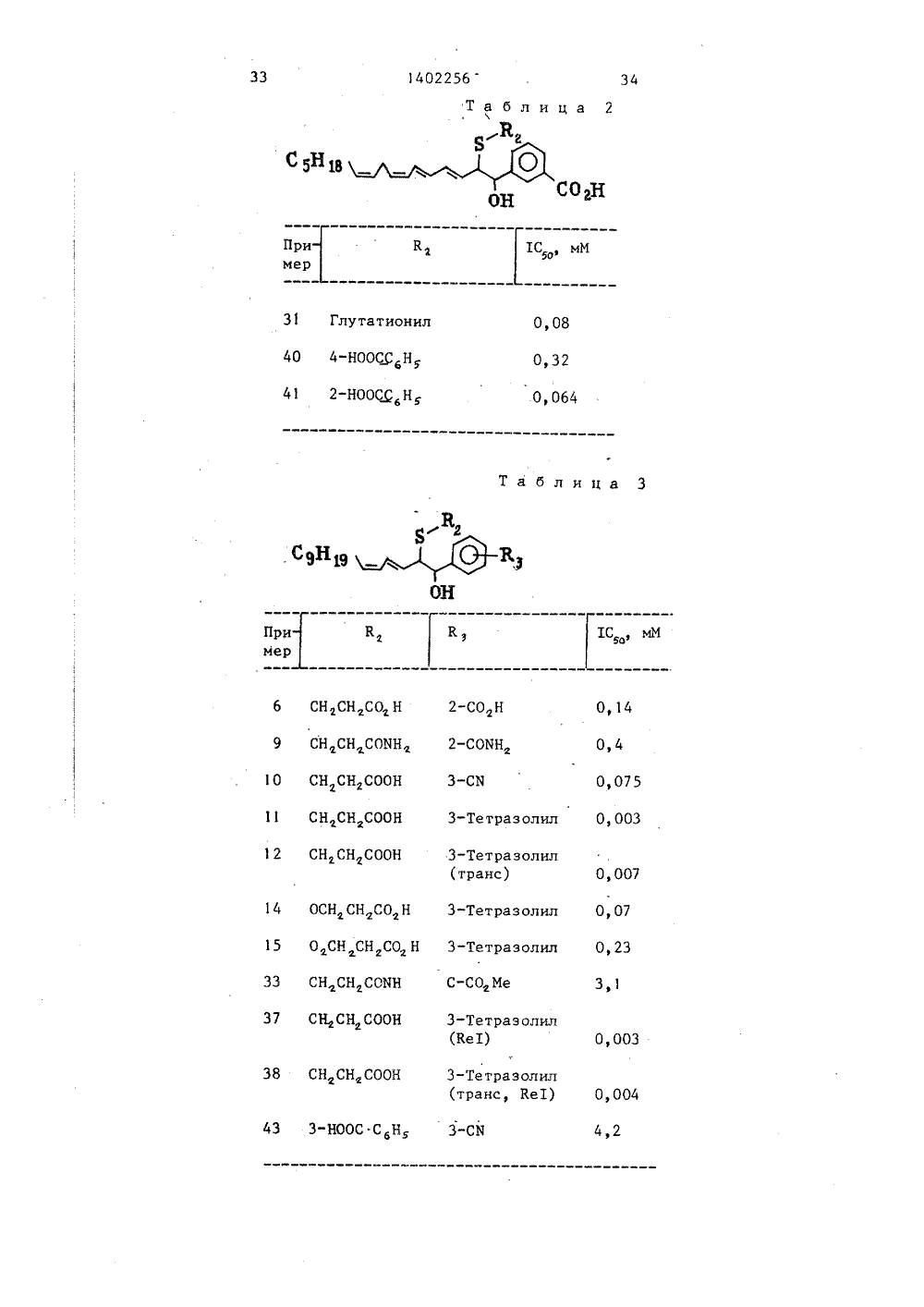
, 140225 ОПИСАНИЕ ИЗОБРЕТЕНИЯН ПАТЕНТУ 1 й 37 77634/23-041.08.84832094303.08.83СВ07.06,88. Бюл. У 21Лилли Индастриз Лимитед(54) СП ОРГАНИЧ (57) Из получен ких соед 0 ГОСУДАРСТВЕННЫЙ КОМИТЕТ СССРпО ДелАм изОБРеТений и ОтнРытий(53) 547.269 А 7 (088.8)56) Вейганд-Хильгетаг. Методы эксперимента в органической химии.М.: Химия, 1968, с. 593. СОБ ПОЛУЧЕНИЯ СЕРОСОДЕРЖАЩИ СКИХ СОЕДИНЕНИЙ ИЛИ ИХ СОЛЕ бретение относится к способ я серососодержащих органиче инений формулы С 07 С 147/02, С 07 С 147/14С 07 С 149/14//А 61 К 31/10 где К, - незамещенный или замещенньфенилом С-С, -алкилен, содержащий 2 4двойные связи; К - фенил, замещенный карбокси-, низшей алкилкарбоксиили цианогруппой, или С,-С, -алкил,который может быть замешен карбокси-,низшей алкилкарбокси-, низшей карбоалкил-, циано-, амидо-, низшей алкиламидо-, тетразолил-, цистеинил-, цистеинилглицинил- или глутанилгруппой;К - карбокси-, циано-, амидогруппаили незамещенный, или замещенныйтрифенилметилгруппой тетразолил; и==0,1 или 2, которые обладают антагонистическим действием, Получение целевых соединений ведут иэ соответствующего оксида и тиола формулыКСИ, где К указано выше, в метанолев присутствии триэтиламина при комнатной температуре с последующимв случае необходимости окислениемполученного соединения формулы (Т),где п=0, до соединения формулы (1),где п=1 или 2, и выделением целевого продукта в свободном виде или ввиде соли. 4 табл.02256 8 1 О 15 20 25 30 35 40 45 50 55 17 14Е. (1 Б, 2 Б)-3 (1,2-0 ксидоб-фенилгексадека(Е), 5 (Е), 13 (2) -триенил) бензойная кислота, сложный метиловый эфирРаствор продукта стадии 0 (1,9 г)и трифенилфосфин (1,3 г) н ацетонитриле (20 мл) нагревают с дефлегмацией н течение 48 ч. Раствор выпаривают, а остаток промывают простымэфиром, затем нысушивают путем добавления бензола и повторного выпаривания с целью получения в осадке неочищенной соли фосфония в виде полутвердой массы,н-Бутиловый литий (1,5 М раствор нгексане, 1,5 мл) добавляют к перемешанному раствору этой соли фосфонияв сухом тетрагидрофуране (50 мл) прио-70 С в азотной атмосфере, Растворярко-желтого цвета перемешивают н теочение 30 мин при -70 С, после чегодобавляют к нему раствор (1 Б, 2 Б)-3(4-формил,2-оксидобут(Е)-енил)бензойной кислоты, сложный метиловыйэфир, в тетрагидрофуране. Смеси дают возможность нагреться до комнатной температуры, выпаривают, а остаток экстрагируют с помощью смесипростого эфира и дихлорметана н соотношении 4:1. Экстракт снова выпаривают, а остаток подвергают хроматографии на силикагелевой колонке, элюируя его с помощью смеси простогоэфира и гексана в соотношении 1:1, сцелью получения соединения в видебледного масла,Р. (1 Б, 2 К)-3-Г 2-(2-Каобоксиэтилтио)-1-окси-фенилгексадека(Е),5(2), 13(Е)-триенил бензойная кислота.Укаэанное соединение получают изпродукта стадии Е примера 3.П р и м е р 30. А, 11-Фенилундеканол.Раствор 11-фенил-ундеценола(2,9 г) в этаноле (300 мл) подвергают гидрированию при 4,22 кгс/см над107-ным палладием на древесном угле(0,6 г) в течение 1 ч, Катализаторпрофильтровывают и фильтрат выпаривают с целью получения целевого соединения в виде бесцветного масла.В, 11- Фенилундеканолтозилат.4-Толуолсульфонилхлорид (2,6 г)добавляют почастям к перемешанномураствору продукта стадии А (2,9 г)ов пиридине (10 мл) при 0-5 С. Раствор перемешивают н течение 16 ч при о0-5 Сзатем слинают в ледяную соляную кислоту и экстрагируют с помощьюпростого эфира. Экстракт промываютрастнорами бикарбоната натрия и хло-,ристого натрия, высушивают и выпаривают. Остаток подвергают хроматографии на силикагелевой колонке, элюируя смесью дихлорметана и гексана всоотношении 1:1, с целью полученияцелевого соединения в виде бесцветного масла,С, (1 Б, 2 К)-3- 2-(2-Карбоксиэтилтио)-1-окси-фенилгексадека(Е),5(Е) -диенил 1 бензойная кислота.Указанное соединение получают изпродукта стадии В с помощью процессов; описанных в примерах 29 (стадия Е) и 3,Прим е ры 31 и 32, (1 Б, 2 К)3- 2-Б-Глутатионил-оксипентадека(Е), 5 (Е)-диенил 1 бензойная кислота и ее 3(Е), 5 (Е)-изомер,Глутатион (300 мл) растворяют всмеси сухого метанола (3 мл) и триэтиламина (1 мл) и добавляют раствор(1 Б, 2 Б-З-(1,2-оксидопентадека(Е),5(2)-диенил)бензойной кислоты, сложный метиловый эфир, н азотной атмосфере, Полученному раствору дали возможность отстояться при комнатнойтемпературе в течение б ч и растворвыпаривают до сухости, К остатку добавляют 2 М раствор гидрата окисилития (3 мл) и раствор перемешиваютв азотной атмосфере в течение 3 чпри комнатной температуре. рН раствора затем доводят до 4 с помощью уксусной кислоты, экстрагируют простымэфиром с целью удаления не полярныхпримесей, Водный растнор подвергают5 раз экстракции с помощью смесихлороформа и метанола в соотношении1:1. Собранные экстракты выпариваютдо сухости с целью получения сыройсмеси целевых соединений, которыеразделены на препаративной противоточной колонке для высококачественной жидкостной хроматографии, элюируемой с помощью смеси метанолаи воды в соотношении 70;30, содержащей в качестве буфера уксусную кислоту и 0,88 аммиака, с целью доведения раствора до рН 5,3. Целевыесоединения представляют собой аморфные твердые вещества бледно-желтогоцвета.П р и м е р 33. Ке 1- 1 К, 2 Б/-3- 2-Б"цистеинилоксипентадека(Е), 19 14022 5(2)-диенил бензойная кислота, сложный метиловый эфир.Раствор 3-(1,2-оксидопентадека(Е), 5(Е) -циенил) бенэойной кислоты, сложный метиловый эфир (0,5 г), сложный И-трифторацетилцистеинметиловый эфир (0,4 г) и триэтиламин (0,5 мл) в сухом метаноле (2,0 мл) подвергают выдержке при комнатной температуре в 10 течение 3 дней и затем выпаривают. Остаток подвергают хроматографии на силикагелевой колонке, элюируя его сначала смесью простого эфира и гексана в соотношении 1:1 и затем прос тым эфиром, с целью получения совершенно защищенного варианта целевого соединения в виде бледного масла,Раствор этого соединения 0,4 г в метаноле (7 мл) и 2 М раствор би карбоната натрия (4 мл) разбавляют водой с целью получения незначительной мутности, а затем выдерживают при комнатной температуре в течение 30 ч. Раствор разбавляют водой 25 (20 мл), подкисляют до рН 4 и экстра. гируют дихлорметаном (Зк 15 мл), Экстракт выпаривают, остаток подвергают хроматографии на силикагелевой колонке, элюируя его смесью дихлорметана и метанола в соотношении 1:1, с целью получения целевого соединения в виде бледного масла.П р и м е р 34. А. 3-(6-Формил,2-оксидогекса(Е), 5(Е) -диенил)35 бензойная кислота, сложный метиловый эфир. Раствор 3- (2-формил, 2-оксидоэтил бензойной кислоты, сложного метилового эфира (0,7 г), в дихлорметане (10 мл) добавляют свьппе 1 ч к перемешанному раствору трифенилфосфоранилиденкротонового альдегида (1,5 г) в дихлорметане (10 мд). Раст 45 вор перемешивают в течение 1,5 ч и затем выпаривают, а остаток экстрагируют простым эфиром. Экстракт выпаривают, а остаток подвергают хроматографии на силикагелевой колон 50 ке, элюируя его простым эфиром, с целью получения масла бледно-желтого цвета, содержащего целевое соединение в смеси с 3(2), 5(Е)-изомером.Раствор этой смеси (230 мг) и иода (10 мг) в дихлорметане (20 мл) перемешивают в течение 2 ч при комнатной температуре и затем выпарива" ют, Остаток промывают гексаном с це 20лью удаления иода, получив целевоесоединение в виде желтого масла.В, 3-(1,2-0 ксидогексацека(Г),5(Е), 10(2)-тетраенил)бенэойная кислота, сложный метиловый офир.н-Бутиловый литий (1,5 М растворв гексане, О,5 мл) медленно добавляют к перемешанному раствору 3-8 ноненилтрифенилфосфонийтозилата(5 мл) при -70 С. Темно-оранжевыйкоричневый раствор перемешивают вотечение 10 мин при -70 С, после чегодобавляют раствор продукта стадии А(210 мг) в тетрагидрофуране (2 мл) .Смесь перемешивают в течение 15 минопри -70 С, позволяют нагреться докомнатной температурь 1 и выпаривают.Остаток подвергают хроматографии насиликагелевой колонке, элюируя егосмесью простого эфира и гексана всоотношении 1:1, содержащей %-ныйтриэтиламин, дополнительно подвергают очистке с помощью высококачественной жидкостной хроматографии с получением целевого соединения в видебледного масла.С. Ке 1-(12,2 Я)-3-2-(2-Карбоксиэтилтио)-1-оксигексадека(Е), 5(Е),7(Е), 10(Е) -тетраенил бензойная кислота, сложный диметйловый эфир.Раствор продукта стадии В (3 мг),метил-меркаптопропионат (2,4 мкл)и триэтиламин (5 мкл) в сухом метаноле (100 мкл) выдерживают при комнатной температуре в течение 3 ч,после чего целевое соединение выделяют с применением высококачественной жидкостной хроматографии. МБМ486.О, Ке-(1 К, 28)-3-2-(2-Карбоксиэтилтио)-1 -оксигексадека - 3(Е), 5(Е),7(2), 1 0(2)-тетраценил 1 бензойная кислота,Раствор продукта стадии С (2,2 мг)в метаноле и 0,5 М раствор карбонатакалия выдерживают при комнатной температуре в течение 16 ч, после чегоцелевое соединение выделяют с помощью противоточной высококачественнойжидкостной хроматографии.П р и м е р 35, А. 3-Меркаптопропионамид.Метил 3-меркаптопропионат (1,2 г)растворяют в 0,88 аммиака (75 мл)и раствор перемешивают при 40 С вазотной среде в течение б ч. Раствор выпаривают до сухости и получен 21 14022 ное твердое вещество белого цвета повторно растворяют в дихлорметане, при этом этот раствор промыт 2 М водной соляной кислотой (10 мл) и высушен (Мд 80)Этот раствор после выпаривания приводит к получению целевого соединения в виде белых пластин, т. пл. 105 С, которые проо,мыты простым эфиром. 10В. (18, 2 К)-3-2-(2-Карбаметилтио) -1-оксипентадека(Е), 5 (Е) -диенилбензойная кислота, сложный метилоный эфир.3"Меркаптопропиоамид (12 мг) раст воряют в сухом метаноле (200 мкл) в азотной атмосфере и добавляют к раствору триэтиламин (100 мкл). Этот раствор добавляют к (18, 28)-3-(1,2- оксидопентадека(Е), 5(Е)-диенил) бензойной кислоте, сложному метиловому эфиру, и полученной смеси предоставляют вазможность отстояться при 40 С в течение 3 ч. Раствор выпариовают до сухости и остаток подвергают 25 хроматографии на силикагелевой колонке, элюируемой этилацетатомЦелевое соединение получено в виде кристаллово белого цвета, т. пл. 65 - 67 С.С. (18, 2 К)-3-12-(2-карбаметилтио) -1-окс ипентадек а(Е), 5 (Е) -диенилбензойная кислота.Раствор продукта из стадии В (40 мг) в тетрагидрофуране (2 мл) и 1 М растворе гидрата окиси лития (0,2 мл) перемешивают в течение 16 ч35 при комнатной температуре. Далее к содержимому добавляют 1 М раствор гидрата окиси лития 0,2 мл и раствор дополнительно перемешивают в те чение 24 ч, после чего разбавляют водой, подкисляют до рН 3 и экстрагируют дихлорметаном, Экстракт высушивают и выпаривают, а остаток дополнительно подвергают очитке путем препа-45 ративной противофазной высококачественной жидкостной хроматографии с целью получения целевого соединения.П р и м е р 36. Ке 1-(1 К, 28)-3- 2-(2-карбоксиэтилтио)- -оксипентадека-З(Е), 5(Е)-диенилбензойная кислота.Продукт примера 3 (стадия В) привысококачественной жидкостной хроматографии показывает содержание приблизительно 107 второго компонента.Выделение этого небольшого по содержанию компонента путем обратно-фазной высококачественной жидкостной 2256хроматографии на колонке, заполненной нуклеозилом С,я, элюируемой смесью метанола и воды в соотношении 80;20, доведенной до рН 5,3 с помощью уксусной кислоты и аммиака, приводит к получению целевого соединения в виде кристаллического твердого вещества,П р и м е р 32, А, 3-Цианокоричный альдегид.Суспензию из активной двуокиси марганца 20 г в растворе 3-цианоциннамилового спирта (4,0 г) в дихлорметане (100 мл) перемешивают при комнатной температуре в течение 16 ч. Смесь профильтровывают и фильтрат выпаривают с получением целевого соединения в виде твердого вещества белого цвета, т, пл. 100 СВ, 3(З-Цианофенил)-1,2-оксидопропанол,Раствор 3-цианокоричного альдегида(2,0 г) в метаноле (20 мл) по каплям добавляют к перемешанному раствору бикарбоната натрия (2,0 г) и507-ной перекиси водорода (1,0 мл) вводе (10 мл). Раствор перемешивают втечение 3 ч при комнатной температуре и затем экстрагируют дихлорметаном. Экстракт высушивают и выпаривают с получением бесцветного масла,которое в основном представляет полуацеталь целевого альдегида,С. 5-(З-Цианофенил)-4,5-оксидо 2-пентенал.Смесь продукта стадии В и формилметилентрифенилфосфорана (3,0 г) вбензоле (150 мл) перемешивают прикомнатной температуре в течение 2 ч,после чего отфильтровывают, Фильтратвыпаривают, а остаток экстрагируютпростым эфиром. Экстракт вновь выпаривают, а остаток подвергают хроматографии на силикагелевой колонке,элюируя смесью простого эфира и гексана в соотношении 3:1, с целью получения целевого соединения в видебледного масла.О. Ке 1-(1 К, 28)-3-(1,2-оксидопен-.тадека(Е), 5(2)-диенил)бензонитрил.Указанное соединение получено изпродукта стадии С с помощью способа, описанного в примере 1 (стадия П).Е. Ке 1-(1 К, 28)-3-12-(2-Карбоксиэтилтио)-1-оксипентадекь(Е), 5 (Е)диенил бензонитрил, сложный метиловыйэфир.23 14Указанное соединение получено изпродукта стадии 1) с помощью способа,описанного в примере 3 (стадия А),Е. Ке-(1 К, 2 Б)-3- 12-(2-Карбоксиэтилтио) - 1-оксипентадекаЗ(Е), 5(2) -диенил бензонитрил.Указанное соединение получено изпродукта стадии Е с помощью способа,описанного в примере 3 (стадия В),С, КеТ-(1 К, 2 Б)-5-3- 12-(2-карбоксиэтилтио)-1-оксипентадека-З(Е),5(2)-диенилфенил - 1 Н-тетразол.Смесь продукта операции Р (100 мг)хлористого аммония (1,0 г) и азиданатрия (1,О г) в диметилформамидео(50 мл) и экстрагируют дихлорметаном.Экстракт выпаривают с получением масла темного цвета, содержащего целевоесоединение и его 3(Е), 5(Е)-изомер всоотношении 40:60, Изомеры разделеныпутем препаративной противофазнойвысококачественной жидкостной хроматографии (на силикагелевой колонке1 Р, -ОДБ, элюируемой смесью метанола,воды и уксусной кислоты в соотношении 85;15:0,1) с целью получения целевого соединения в виде кристаллического твердого вещества, т. пл.153 - 155 С. МБ (ББА)М 473,П о и м е р 38. Ке-(1 К, 2 Б)5- 3-12- (-2-карбоксиэтилтио)-1-оксипентадека(е), 5 (е)-диеннл фенил -1 Н-тетразол.Указанное соединение отделяют отпродукта реакции, описанного в примере 37 (стадия 0),П р и м е р 39. А. (1 Б, 2 К)3- 2-(3-Метоксикарбонилфенилтио) в 1 оксипентадека-З(Е), 5(2)-диенил 1 бензойная кислота, сложный метиловыйэфир,К раствору метил-меркаптобензоата (0,42 г) в метаноле (2 мл) вазотной атмосфере добавляют триэтиламин (0,38 мл) с проявлением слабожелтой окраски, Смесь затем переливают в другую колбу, содержащую (1 Б,2 Б)-3- 1,2-оксидопентадека-З (Е), (52)диенилбензойную кислоту, сложный метиловый эфир (0,8 г), в азотной атмосфере, Реакционную смесь перемешивают при комнатной температуре вазотной атмосфере в течение 2 ч.Летучие удалены с помощью азотного потока, а остаток очищают с по 02256 24мощью колоночной хроматограФии (двуокись кремния; растворитель для элюирования - 507-ная смесь гексанз ипростого диэтилового эфира) с целью5получения продукта в виде светложелтого масла, Протонный ЯМР указывает на преимущественную (Е), (2)- стереохимию (пространственное строе 10 нне)В. (1 Б, 2 К)-3- 2-(3-Карбоксифенилтио) - 1-оксипентадека(Е), 5 (2)-диенил бенэойная кислота,К раствору эфира двухосновной кислоты стадии А (0,46 г) в тетрагидрофуране (2 мл) добавляют 2 Мраствор гидрата окиси лития (2,6 мл),Двухфазную систему энергично перемешивают в течение 20 ч при комнатнойтемпературе,Тетрагидрофуран удаляют под вакуумом, а водную фазу осторожно подкисляют до рН 4 с помощью 2 М соляной кислоты. Экстракция с помощью25 хлороформа с последующей сушкой(сульфат магния) и выпаривание приводят к получению соединения в видетвердого вещества светло-желтогооцвета, т. пл. 90 С (с смолообразованием) . П р и м, е р ы 40-42. Аналогичнопримеру 39 получены следующие соединения(1 Б, 2 К)-3- 2-(4-карбоксифенил тио) - 1-оксипентадека - 3 (Е), 5 (2)-диенил 1 бензойная кислота (клейкое твердое вещество желтого цвета);(1 Б, 2 К) - .3-12- (2-карбоксифенилтио) - 1-оксипентадека(Е), 5 (2)-дие нилбензойная кислота клейкое твердое вещество);(1 Б, 2 К)"3- 2-бутилтио - 1-оксипентадека-З(Е), 5(2)-диенилбензойнаякислота,45Пример 43. А, (1 Б, 2 К) 31-(3-Цианофенил) -1-оксипентадека 3 (Е), 5 ( 2 ) -д Фен-илт Фо б ен з ой на якислота, сложный метиловый эфир.50Краствору метил-меркаптобензоата (0,29 г) в метаноле (1 мл) вазотной атмосфере добавляют триэтиламин (0,26 мл) с проявлением желтой 55 окраски, Смесь добавляют к (1 Б, 2 Б)- 3-(1-оксидопентадека-З(Е), 5(2)-диенил)бензонитрилу (0,5 г) в азотнойсреде, реакционную смесь перемешива 402256 26ют при комнатной температуре в течение 4 ч.Летучие удалены с помощью потокаэота а остаточное масло очищают спомощью хроматографии (двуокись кремния); растворитель для элюированиямесь гексана и простого эфира) селью получения продукта в виде желого масла,В. (18, 2 К)-3- 1-(3-Цианофенил)1-оксипентадека(Е), 5 (2)-диенлтио 1 бензойная кислота.К раствору сложного метиловогофира из стадии А (50 мг) в тетрагидофуране (0,3 мл) добавляют 2 М растор гидрата окиси лития (0,15 мл)месь перемешиваютпри комнатнойемпературе в течение 24 ч.Тетрагидрофуран выпаривают подакуумом, водную фазу подкисляют 2 Моляной кислотой, дважды экстрагируютпомощью дихлорметанаСушка (с поощью сульфата магния) и выпариваниерганических экстрактов приводят колучению целевого соединения в видеветло-янтарного масла ( протонныйР и противофазная высококачественая жидкостная хроматография покаьвают на наличие приблизительно 30%(Е), (Е)-изомера),П р и м е р 44. А. 28-Оксидо-фенилпропиональдегид.К 507.-ному раствору перекиси водорода (16 мл), к которому в качестве буферного раствора добавленнасыщенный раствор бикарбоната натрия, по каплям добавляют с охлаждением в ледяной ванне раствор коричногоальдегида (26,4 г) в метаноле (100 мл) .Смесь перемешивают при комнатнойтемпературе в течение 4 ч.Метанол удаляют под вакуумом, аводную фазу дважды экстрагируют спбмощью толуола. Высушенные экстракты использованы без выделения целевого соединения,В. 4,5-0 ксидо-фенил-пентанал,Раствор 2,3-оксидо-фенилпропиональдегида в сухом толуоле (300 мл)обраба тьв ают формилметилентрифенилфосфораном (60,8 г) и смесь перемешивают при комнатной температуре втечение 20 ч; .Реакционную смесь отфильтровывают и выпаривают под вакуумом, а остаточное твердое вещество трижды экстрагируют простым эфиром с применением ультразвуковой ванны. Экстракты простого эфира профильтровывают ивыпаривают с получением масла, которое далее очищают с помощью колоночной хроматографии (двуокись кремния: разбавитель для элюированиясмесь гексана и простого эфира в соотношении 1:1) с целью получения 10 продукта в виде желтого масла.С. 1,2-0 ксидофенилпентадека(Е),5(2)-диен.К раствору бромистого н-децилтрифенилфосфония (20,54 г) в сухом тетрагидрофуране (200 мл), охлажденного до -70 С (ванна из сухого льда иацетона), в азотной атмосфере добавляют н-бутиловый литий (26,6 мл 1,5 Мраствора гексана), При этом сразупоявляется оранжевая окраска, Смесьперемешивают в течение 1 О мин, после чего к ней добавляют раствор 4,5 оксидо-фенил-пентанала в сухомтетрагидрофуране. Реакционной смеси 25 дают воэможность нагреться до комнатной температуры и перемешивают в течение 2 ч.Тетрагидрофуран выпаривают подвакуумом, остаточное полутвердое веЗ 0 щество экстрагируют 4 раза простьмдиэтиловым эфиром с использованиемультразвука, а экстракты профильтровывают и выпаривают с получением желтого маслаЭто масло затем очищают 35с применением хроматографии (двуокиськремния; растворитель для элюирования - смесь гексана и воды в соотношении 1:1) с целью получения продукта в виде подвижного желтого масла,которое кристацлизуется при охлаждениидо -20 С, т, пл, ( 50 С.О, Ке 1-(1 К 25)-2-(2-метоксикарбонилэтилтио)-1-окси-фенилпентадека-З(Е), 5(Е)-диен.45К раствору метил-маркаптопропионата (044 г) в метаноле 2 мл вазотной атмосфере добавляют триэтиламин (0,5 мл). Смесь переливают вколбу, содержащую эпоксид стадии С(1,00 г) в азотной атмосфере, Реакционную смесь перемешивают в течение20 ч, затем дополнительно добавляюттиол (0,44 г) и триэтиламин (0,5 мл)..При выдержке в условиях комнатнойтемпературы 20 ч летучие удаляют в 55 потоке азота, а остаток подвергаютколоночной хроматографии (двуокиськремния; растворитель для элюирования - смесь гексана и простого ди 27 140этилового эфира в соотношении 3:1)с получением целевого соединения ввиде бесцветного масла,Е, Ке 1-(1 К, 2 Я)-2-(2-карбоксиэтилтил)-1-окси-фенилпентадека(Е),5(2)-диен.Смесь сложного метилового эфираоперации Д (0,49 г) в тетрагидрофуране (3 мл) и 1 М раствор гидрата окиси лития (3,5 мл) перемешивают прикомнатной температуре в течение24 ч,Тетрагидрофуран выпаривают подвакуумом, водную фазу подкисляют 2 Мсоляной кислотой, дважды экстрагируют простым диэтиловым эфиром, Собранные органические экстракты высушивают (с помошью сульфата магния) ивыпаривают с получением целевогосоединения в виде янтарного масла,П р им е р 45 (альтернативныйметод). А. 3-3-(2-ТрифенилметилНтетразол-ил)фенил-пропенол.К раствору 3-3-(1 Н-тетразол-ил)фенил 1-2-пропенола (2,02 г) в сухомдихлорметане (50 мл) добавляют триэтиламин (1,5 мл), а затем трифенилхлорметан (2,8 г) в сухом дихлорметане. Раствор перемешивают при комнатной температуре в течение 90 мин,промывают водой (50 мл), затем раствором бикарбоната натрия (57-ный,50 мл), высушивают над сульфатом магния, отфильтровывают и выпаривают подпониженным давлением с целью получения бледно-коричневого вязкого масла,которое подвергается кристаллизациипри отстое до латексного состояния.В. (2 Я, ЗЯ) -3- 3-(2-ТрифенилметилН-тетразол-ил)фенил -2,3-оксидопропанол.2-(+)-Диметиловый тартрат (1,85 г)по капле добавляют в сухой дихлорметан (1 О мл) к перемешанному растворуизопропокси титана (117) (3,1 мл ) всухом дихлорметане (30 мл) при -20(6,7 мл), оба при температуре -20а-25 СРаствор бледно-оранжевого цвета оставляют в холодильнике на 3 ч.К перемешанному раствору добавляютводный раствор винной кислоты(107,-ный, 50 мл) и смесь перемешивают н течение 1 ч, отфильтровывают и разделяют. Лихлорметановый слой высушен над сульфатом магния, отфильтрован и выпарен под пониженным давлением с получением желтого масла. Масло растворяют в четыреххлористом углероде, промывают водой, высушивают над сульфатом магния, отфильтровывают и выпаривают под вакуумом с целью получения бледно-желтого масла, Масло подвергают хроматографии на силикагелевой колонке с использованием смеси простого диэтилового эфира и гексана в соотношении 2:1, необходимые фракции выпаривают при пониженном давлении с целью получения бесцветного кристаллического твердого вещества,С. (4 Я, 5 Я) -5-13-(2-Трифенилметил 2 Н-тетразол-ил)фенил,5-оксидо-пентенал.Твердую трехокись хрома (5,0 г) добавляют к перемешанному раствору пиридина (7,9 мл) в сухом дихлорметане (200 мл) при 5 С. Смесь перемешиваюто в.течение 45 мин, подогрев до 13 С, и, позволив всей трехокиси хрома раствориться, быстро вводят раствор эпоксиспирта стадии В (4,61 г) в сухом дихлорметане (50 мл). Темную смесь перемешивают в течение 90 мин, нагревая до комнатной температуры, после чего профильтровывают через Флоризилоную прокладку с целью удаления солей хрома, бесцветный фильтрат выпаривают под давлением с получением бледно-желтого масла.К раствору масла (1,8 г) в бензоле (75 мл) н азотной атмосфере добав" ляют в виде одной порции формилметилентрифенилфосфоран 1,34 г),Суспензию перемешивают при комнатной температуре в азотной атмосфере в течение 8 ч, непрореагировавший продукт отфильтронан, а фильтрат выпаривают под пониженным давлением до коричне" ного масла, Последнее экстрагируют горячим простым эфиром, охлаждают, отфильтровывают и выпаривают под пониженным давлением с получением желтого масла, которое подвергается при отстаивании кристаллизации с образованием твердого вещества желтого цвета. О, (1 Я, 2 Я) -5- 3- 2- (1, 2-Ок с идо) пентадека(Е), 5 (2)-диенилфенип) - 2-трифенилметилН-тетразол.02256 30 29 4н-Бутиловый литий (8,91 мл, 1,5 М)в гексане по капле добавляют к перемешанному раствору бромистого н-децилтрифенилфосфония (6,07 г), высуощенного при 80 С под пониженным давлением в течение 16 ч, в сухом тетОрагидрофуране 130 мл при -70 С, вазотной атмосфере. Прозрачный ярко оранжевый полученный раствор перемео; после чего к нему по капле добавля ют раствор 5- 3-(2-трифенилметилН 1 тетразол-ил)фенил 1-4,5-оксидопентенала (6,4 г) в сухом тетрагидрофуране (75 мл), Бледно-желтый раствороперемешивают при -70 С в течение 1.ч,дают возможность нагреться до комнат ной температуры и выпаривают под пониженным давлением до коричневогомасла. Масло экстрагируют смесьюпростого эфира и гексана (в соотношении 1:2, 4 ч 200 мл) и бледный мут, ный экстракт выпаривают под понижен-(4,5 г) и триэтиламина (2,06 мл) вметаноле (15 мл) помещают в колбу, ватмосфере азота, Этот раствор добавляют к метил"тиопропионату (900 мг)в атмосфере азота и раствор перемешивают в течение 24 ч при комнатнойтемпературе до полного завершения реакции. Раствор выпаривают под пониженным давлением с получением в остатке коричневого масла, которое подвергают хроматографии на кодонке, заполненной двуокисью кремния, с использованием смеси простого эфира игексана в соотношении 1:1, Требуемыефракции выпарены под пониженным давлением с получением целевого соединения в виде желтого масла.Р. (1 Я, 2 К)-5-(3-2-(2-Карбоксиэтилтио) -1-оксипентадека(Е), 5 (2)- диенил 1 фенил -1 Н-тетразол, натриеваясоль,115, 22)-5-(3-(2-Метоксикарбоиилэтилтио) -1-оксипентадека(Е), 5 (Е)- диенил 1 фенил) -2-трифенилметилН-тетразол (2,74 г) растворяют в простомэфире (50 мл), к которому добавля 5 10 15 20 25 30 35 40 45 50 55 ют водный раствор соляной кислоты (20 мл; 5 М) и смесь перемешивают при комнатной температуре в течение 4 ч, пока тонкослойная хроматография не показывает, что потеря трифенилметильной зашиты завершена. Простой эфир выпаривают пад пониженным давлением, затем добавляют тетрагидрофуран (30 мл) с последующим введением водного гидрата окиси лития (2 М) до тех пор, пока раствор не стал щелочным. Смесь оставляют под перемешиванием на ночь при комнатной температуре. Водную основную фазу отделяют, подкисляют водной соляной кислотой (2 М) экстрагируют простым эфиром (2 ю 50 мл) и эфирный экстракт выпаривают под пониженным давлением с получением коричневого масла. Масло подвергают хроматографии на колонке, заполненной двуокисью кремния, с помощью простого эфира, необходимую фракцию выпаривают под пониженным давлением с целью получения бледно- желтого масла. Масло растворяют в водном бикарбонате,натрия (0,5 М;1 экв.) и высушивают при отрицательной температуре с получением натриевой соли.Соединения изобретения являются фармакологически активными, будучи антагонистами лейкотриена, как видно при испытаниях п чуго на сегментах кишечника морских свинок при концентрациях от 10 нг до 50 мкг. Согласно способу Шилда.незащищенные соединения формулы (1), описанные в табл, 1 - 4, демонстрируют 1 С против ЕТ 1)4 менее чем 10М. Соединения изобретения также активны при функциональных легочных опытах над морскими свинками п чиччо Остена и Дрейзена при уровнях,внутривенной дозировки от 0,05 мкгдо 6,0 мг/кг и в видоизмененном опыте Герксхеймера при дозировках от 25 до 200 мкг/кг. Опыт Герксхеймера основан на бронхоспаз 4 ме у морских свинок, вызванном ЕТД который напоминает астматическое воздействие на человека.При приготовлении композиций, предусмотренных изобретением, активный ингредиент обычно смешивают с носителем или разбавляют носителем, и/или помещают в носитель.Соединения формулы (1) являются низкотоксичными и обладают свойства-. ми лейкотриеновых антагонистов,32 Таблица 1 ОН С, м 50 ри ер К К ис те инил (Ке 1) теинилглици 3 и о 2 теин 16 СН -СН -тетразо лил 0,005 ои; гр К -ка Эси-, циан или нез ид 0,0 мещенныйрифенилмзолил; групили)и ме 0 илгруппои те ыих со или 2,отличнение общ,4 ющийс яформулы что сое 7 22 0,4 еК,идвергаюрмулы,4 ф 7 о 2 СН )СО 11 НСН СО начен Н СН ламипо 7 димости 0 диненениясоединениили 2, и 39 ои-бути и 31 1402256Формула изобретения Способ получения серосодержащих органических соединений общей формулы ОН5 незамещенный или замещенныйфенилом С-С,-алкилен, со"держащий 2-4 двойные связи;фенил, замещенный карбоксинизшей алкилкарбокси- илицианогруппой, или С,-Сю -алкил, который может бытьзамешен карбокси-, низшейалкилкарбокси-, низшей карбоалкил-, циано-, амидо-,низшей алкиламидо-, тетразолил-, цистеинил-, цистеинилглицинил- или глутанилимеют указанные значениаимодействию с тиолом где К имеет указанные з в метаноле в присутствии на при комнатной темпера следующим в случае необх окислением полученного с формулы (1), где п=О, д общей формулы (1), где и выделением целевого прод бодном виде или виде сол СН(СН ) СО, НСН(СН 5)СН СОНСН СН(СН. ) СОН1402256 33 34 ри ер 3 утатион 4-НООЫ 0 ОО О, 064 Т 3 л мМ Пр ме 2-СО Н С 2-СОНН О,0,075,00 СН СН СО етразолианс) 0,00 0 Ме Тетразолрансь 0,00 3-СЫ 9 СН СН СОИ 10 СН СН СОО 11 СН СН СОО 14 ОСН СН СОг ОгСНгСНгСОг Н33 СН СНгСОНН 7 СН СН СОО 38 СН СНгСОО 3 3-НООС С,Н,Таблица 2 ВР 3-Тетразолил Тетразолил Э-Тетразолил 3-ТетраэолилИзобретение относится к способу получения серосодержащих органических соединений формулы где ко 35 К, - незамещенный или замешенныйфенилом Сц-С,-алкилен, содержащий 2-4 двойные связи;К - фенил, замещенный карбокси-,низшей алкилкарбокси- илицианогруппой, или С,-С,-алкил, который может быть замещен карбокси-, низшей алкилкарбокси-, низшей карбоалкил", циано-, амидо-, низшей алкиламидо-, тетразолил-, цистеинил-, цистеинилглицинил- или глутанилгруппой;К - карбокси-, циано- амидогруппа или незамещенный,или замещенный трифенилметилгруппой тетразолил;п=0, 1 или 2,торые обладают антагонистическимдействием на рецепторы лейкотриенаи могут быть использованы при лечении аллергических заболеваний,Цель изобретения - разработкаспособа получения новых соединенийформулы (1), которые обладают антагонистическим действием на рецепторы микотриена.П р и м е р 1А. Альдегид 3-карбоксикоричной кислоты, сложный метиловый эфир. 40Сложный метиловый эфир 3-карбоксибензальдегида (16,4 г) растворяют втолуоле (125 мл) и добавляют кристаллический Формилметилентрифенилфосфоран (30,4 г) с перемешиванием вьсреде азота. Смесь доводят до 100 Си перемешивают в течение 4 - 5 ч.Раствор темно-соломенного цвета выпаривают в вакууме и остаток подвергают экстракции с помощью простого 50диэтилового эфира. Нерастворимуюокись трифенилфосфина удаляют путемфильтрации, а фильтрат темно-желтого цвета подвергают концентрированию в вакууме и хроматографированию 55в колонке, заполненной силикагелем,с использованием в качестве проявляющего агента диэтилового эфира /н нилфосфониясухом тетрамид н-децилтриг) растворяют 2,гексана в объемном соотношении 50/50, .Целевое соединение получено в виде бледно-желтого кристаллического твердого вещества, т. пл, 86 - 87 С,В. 3-(2-Формил,2-оксидоэтил) бензойная кислота, метиловый эфир.Сложный метиловый эфир альдегида 3-карбоксикоричной кислоты (400 мг) растворяют в метаноле (12 мл) и по каплям добавляют к перемешанному раствору бикарбоната натрия (640 мг) и 28%-ной перекиси водорода (1,6 мл) в воде (24 мл), После завершения добавления смесь подвергают перемешиванию в течение 1,5 ч при комнатной температуре. В этой фазе жидкостная хроматография показывает отсутствие исходного альдегида, Мутный раствор подвергают экстракции дихлорметаном (Зк 10 мл) и выпаривают в вакууме с получением почти бесцветного масла (453 мг) в основном в виде целевого альдегидгидрата. Азеотропная перегонка этого продукта от бензола приводит к получению целевого соединения в виде почти бесцветного масла (340 мг), которое легко кристаллизируется при хранении в холодильнике.С. 3-(4-формил,2-оксидобут(Е) - енил)бензойная кислота, сложный метиловыи эфирПродукт стадии В растворяют в бензоле (40 мл) и добавляют кристаллический формилметилентрифенилфосфоран (2 г), смесь перемешивают при комнатной температуре в атмосфере азота в течение 2 ч. Бензол выпаривают в вакууме, а остаток экстрагируют с помощью диэтилового эфира с целью удаления нерастворимой окиси трифенилфосфина. Экстракт простого эфира выпаривают и остаток растворяют в небольшом количестве диэтилового эфира/н-гексана в объемном соотношении 50/50, подвергают хроматографии на колонке, заполненной силикагелем, с использованием одной и той же смеси растворителя для проявленияФракции, содержащие целевое соединение, собраны и выпарены с целью получения соединения в виде бледно-желтого масла.Р, 3- (1, 2-Оксидопентадека(Е),5(2)-диенил)бензойная кислота, слоный метиловый эфир.2256а 51 О 15 20 25 30 35 Полученное твердое вещество отфильтровывают и промывают водои до сушкив вакууме при 50 С в течение 2 дней.Сырой продукт затем подвергают перекристаллизации от ледяной уксуснойкислоты с целью получения бесцветныхопластин, т. пл. 189 - 190 С, исключительно Е-изомер.В, Хлористый (Е)-3-метоксикарбонилциннамил,Хлористый оксалил (13,9 г) до 50 3 40гидрофуране (50 мл), перемешивают воатмосфере азота и охлаждают до -78 С.Бутиловый литий (1,55 М раствор вгексане, 3,4 мл) постепенно добавляют с немедленным появлением оранжево-желтой окраски. Через 15 мин ксодержимому быстро добавляют растворпродукта стадии С в тетрагидрофуране (5 мл), раствор выдерживают при-78 С в течение 20 мин, затем полуоченной смеси дают возможность медленно принять комнатную температуру,Раствор выпаривают под вакуумом иостаток экстрагируют с помощью диэтилового эфира /н-гексана в объемномсоотношении 50/50. Экстракт растворителя подвергают концентрированиюи хроматографии на колонке с силикагелем, используя для проявления однуи ту же смесь растворителя. Целевоесоединение получено в виде бесцветного масла, кристаллизующегося призамораживании,Е. Ке 1-(1 К, 2 Б)-3-(2-Я-цистеинил - 1-оксипентадека(Е), 5(Е)-диенил)бензойная кислота.Соединение стадии 0 приведено вовзаимодействие в азотной среде сраствором сложного метилового эфираМ-трифторацетилцистеина (0,88 г) итриэтиламина (1,1 мл)в сухом метаноле (4, 5 мл) при комнатной температуре в течение 24 ч. После этоговремени вся масса исходного эпоксидаисчезает, что подтверждено с помощьюжидкостной хроматографии,Раствор выпаривают под вакуумом,затем растворяют в небольшом количестве простого диэтилового эфира/н-гексана в объемном соотношении50/50 и.подвергают хроматографии вколонке, заполненной силикагелем,проявляясь сначала с помощью той жесамой смеси растворителя, с цельюудаления мельчайших количеств исходного эпоксида, затем с помощью простого диэтилового эфира с получениемполностью защищенного варианта целевого соединения в виде окрашенногомасла слабосоломенного цвета.Продукт растворяют в тетрагидроФуране (7 мл) и добавляют растворгидрата окиси лития (1 М, 8 мл) с последующим добавлением воды для получения мутного раствора. Через 3 дня5гидролиз еще неполон, дополнительнодобавляют раствор гидрата окиси лития (4 мл), После еще 4 дней светлый раствор при рН приблизительно равном 11 подвергают экстракции с помощью простого диэтилового эфира, Остаточную водную Фазу затем тщательно отрегулируют до рН 3,5 - 4 (слабой соляной кислотой), после чего несколько раз подвергают экстракции с помощью дихлорметана /метанола в объемном соотношении 3/. Всю органическую фазу тщательно выпаривают под вакуумом с целью получения целевого соединения, первоначально в виде масла бледно-соломенного цвета, которое постепенно превращается в хрупкое твердое вещество.Соединение Ке 1-(К, 28)-3-(2-8- цистеинилглиницил-оксипентадека 3 (Е) -5 (2) -диенил) бензойная кислота в виде масла светло-соломенного цвета получено аналогичноП р и м е р 2. А. 3-Метоксикарбонилкоричная кислота.Сложный метиловый эфир 3 в карбоксибензальдегида (82 г) растворяют в сухом пиридине (250 мл) и к перемешанному раствору добавляют малоновую кислоту (52 г)Затем добавляют пиперидин (5 мл) и раствор медленно нагревают до дефлегмации. При этомимеет место несколько экзотермическая реакция сопровождаемая выделением углекислого газа, После дефлегмации раствора в течение 1 ч добавляют дополнительное количество малоновой кислоты (25,1 г). Затем растворподвергают дефлегмации в течение30 мин перед охлаждением и добавляют лед и 5 М соляную кислоту (1 л),бавляют к перемешанной суспензии 3-метоксикарбонилкоричной кислоты(20,6 г) в сухом простом эфире(200 мл) и затем для ускорения реакции добавляют 1 каплю ДМФ. После перемешивания в течение 1 ч при комнатной температуре все твердое веществорастворяют и раствор выпаривают досухости с получением белого кристал 1402256 б(24 г) растворяют в сухом тетрагидрофуране и затем этот раствор добавляют к три-трет-бутоксиалюмогидриду лития 63,5 г , растворенному в тетрагидрофуране (250 мл) при -78 С, Полученный светлый раствор перемешиваютопри -78 С в течение 30 мин, затем егодобавляют ко льду и 2 М соляной кислоте (750 мл). Двухфазную смесь 4 раза подвергают экстракции с помощьюдихлорметана, После сушки дихлорметан выпаривают с получением целевогосоединения в виде бледно-желтого масла,П. Сложный метиловый эфир (Е)-3(З-окси,2-оксидопропил)бензойнойкислоты.3-Метоксикарбонилциннамиловыйспирт (1,92 г) растворяют в дихлорметане (50 мл), охлаждают до 0 С и метахлорпероксибензойную кислоту(1,72 г) в 10 мл дихлорметане покаплям добавили к охлажденному раствору. Затем дают возможность температуре реакционной смеси поднятьсядо комнатной температуры, Через 2 чм-хлорбензойную кислоту отфильтровывают и раствор промывают дваждынасьшенным раствором бикарбоната натрия, После сушки и выпаривания слоядихлорметана получено целевое соединение в виде бесцветного масла.Е, (18, 28) 3-(3-Окси - 1,2-оксидопропил)бензойная кислота, сложный метиловый эфир (альтернативный метод),Тетраизопропоксид титана (1,3 мл)растворяют в сухом дихлорметане(12 мл) и перемешанный раствор охолаждают до -60 С. Добавляют 1-диэтилбвый тартрат (6,0 мМ), раствору даоют возможность нагреться до -20 С иперемешивают еще в течение 10 мин.Затем добавляют 3-метоксикарбонилциннамиловый спирт (960 мг). В концедобавляют 3 М раствор гидроперекиситрет-бутила (6 мл) й 1,2-дихлорэтанеи раствор выдерживают при -18 С втечение 16 ч. К реакционной смеси затем добавляют простой эфир (15 мл) споследующим добавлением насыщенногораствора водного сульфата натрия(2 мл). Смесь перемешивают при комнатной температуре в течение 1 ч и(240 мг) растворяют в сухом метаноле3 мл). Затем добавляют триэтиламин45,(250 л) и полученный раствор добавляют к сложному метиловому эфиру 3(1,2-оксидопентадека(Е), 5 Е-диенил)бензойной кислоты (712 мг). Полученный светлый раствор выдерживают при40 С в течение 1 6 ч, после чего выпаривают до сухости и подвергают хроматографии на колонке, заполненнойдвуокисью кремния и элюированнойэфиром, Целевое соединение полученов виде бесцветного масла,В. Ке 1-(1 К, 28)-3-12-(2-карбоксиэтилтио)-1-оксипентадек 3(Е), 5(2)- диенил 1 бензойная кислота. 5 10 15 20 25 30 35 40 отфильтровывают через цеолит. К раствору добавляют толуол (100 мл) и выпаривают его до получения бесцветногомасла, которое подвегают хроматографии в силикагелевой колонке, элюируемой простым эфиром. Диэтил тартрат,элюированный первым, предшествуетцелевому соединению, которое получено после выпаривания растворителя ввиде бесцветного масла,Р. 3-(2-формил,2-оксидоэтил)бензойная кислота, сложный метиловыйэфир.Трехоксиь хрома (2,5 г) добавляют к раствору пиридина (3,9 г) в дихлорметане (100 мл) при 7 СТемпературе перемешанного раствора даютовозмуность подняться до 14 Сдят сложный метиловый эфир 3(3-окси 1,2-оксидопропил бензойной кислоты(1,04 г) в дихлорметане (2 мл), Раствор темнеет и темное масло выделяетсяиз раствораЧерез 30 мин при 22 Сслой дихлорметана декантируют и профильтровывают через Фторозил. Послевыпаривания этого раствора полученоцелевое соединение в виде бесцветного масла.С, Ке 1-(1 К, 28)-3- 2-Б-цистеинил 1-оксипентадека(Е), 5(Е)-диенил 1бензойная кислота.Указанное соединение получают изсоединения стадии Р с помощью способов, описанных в примере 1 (стадииС, П и Е),П р и м е р 3, А, Сложный диметиловый эфир КеТ-(1 К, 28)-3- 2-(2 карбоксиэтилтио)-1-оксипентадека 3-(Е), 5(Е)-диенил 1 бенэойной кислоты,02256 8 7 14Сложный диметиловый эфир Ке 1-(1 К,2 Б)-3-2-(2"карбоксиэтилтио) -1-оксипентадека(Е), 5 (2) -диенил бензойной кислоты (476 мг) растворяют втетрагидрофуране (1 О мл) и 1 М растворгидрата окиси лития (3 мл) добавляют к нему. Смесь перемешивают прикомнатной температуре в течение 2дней, с помощью слабой соляной кислоты рН раствора затем доводят до 4.В конце этот раствор четыре раза экстрагируют с помощью дихлорметана,который после сушки (МрБО 4) и выпаривания приводит к получению целевого соединения в виде твердого небелого вещества, т. пл. 87 - 90 С.оП р им е р 4, А. (1 Б, 2 Б)-3(1,2-0 ксипентадека-З (Е), 5 (2)-диенил)бензойная кислота.Указанное соединение получено изпродукта примера 2 (стадия Е).при помощи способов, описанных в примерах 2(стадия А) с целью получения целевогосоединения в виде бледного маслаМБ (ББА) М 477.С. (1 Б, 2 К)-3-12 ф-Карбоксиэтилтио)-1-оксипентадека(Е), 5 (2) -диенил бензойная кислота.Эфир двухосновной кислоты подвергают гидролизу (пример 3, стадия В)с целью получения соединения в видебелого твердого вещества, т. пл.приблизительно 50 С,о и +50,1(1 К, 2 К)-3-(3-0 кси,2-оксидопропил)бенэойную кислоту, сложный метиловый эфир получают по примеру 2(стадия Е) с помощью Д(-)-диэтилового тартрата вместо 1 (+) изомера.Этот эпоксид затем вводят в реакциюпо примерам 2 (стадия Р), 1 (стадия С и 0), 3 ( стадииА и В) с цельюполучения целевого соединения в виде бледного масла.П р и м е р б. А. (Е)-2-Метоксикарбонилциннамиловый спирт. 5 10 15 20 25 30 35 40 45 50 55 2-Г 1 етоксикарбонилциннамиловыйоспирт,.тпл, 176 С, получен по примеру 2 (стадия А) и превращен в хлоО рид кислоты, т. пл. 75 - 80 С с помощью метода, описанного в примере 2 (стадия В). Раствор этого хлорида (15,5 г) в простом эфире (250 мл) добавляют к перемешанной суспенэии боргидрида натрия на двуокиси кремния (60 г), полученной путем добавления раствора из 1 ч, боргидрида натрия в 2 ч, воды к 10 ч. двуокиси кремния, с охлаждением и последующей сушкой под вакуумом, в простом эфире (360 мл), Смесь перемешивают в течение 2 ч при комнатной температуре и отфильтровывают. Фильтрат промывают 107.-ным водным раствором карбоната натрия, затем насыщенным раствором хлористого натрия, высушивают и выпаривают с получением целевого соединения в виде бесцветного маслаВ, (Е)-2-(З-Окси,2-оксидопропил)бензойная кислота, сложный метиловый эйирОкисление (Е) -2-метоксикарбонилциннамилового спирта хлорпероксибензойной кислотой (пример 2, стадия Э) с последующей хроматографией сырого продукта в колонке, заполненной двуокисью кремния, элюированной смесью простого эфира и гексана в соотношении 2:1, приводит к получению целевого соединения в виде бледного масла,С. 3- 1-2-Метоксикарбонилэтилтио)-тетрадек(Е), 4(2)-диенил 1- 1,3-дигидроизобензофуран-он.Сложный метиловый эфир 2-(4-Формил,2-оксидобут-З(Е)-енил)бензойной кислоты, т. пл.50 С, получен иэ соединения стадии В с помощью процессов, описанных в примерах 2 (стадии Э и Р) и 1 (стадия С), Последующая реакция этого соединения способами, описанными.в примерах 1 (стадия О) и 3 (стадия А), приводит к получению основного продукта целевого соединения лактона.П. Ке 1-(1 К, 2 Б)-22-(2-карбоксиэтилтио) -1-оксипентадека(Е), 5 (2) - диенил бензойная кислота, двунатриевая соль.Раствор 3-1 в (2-метоксикарбонилэтилтио)-тетрадека(Е), 4(2)-диенил - 1,3-дигидроизобензофуран-он (115 мг) в тетрагидрофуране (0,96 мл) перемешивают при комнатной темпера40 8. А. 1-Бромо-ме 55 9 14022туре в течение 16 ч. Раствор выпаривают и остаток промывают эфиром сцелью выделения целевого соединенияв виде вязкой смолы,П р и м е р 7. А. 9-Бромо-ме 5тил-децен,н-Бутиловый литий (1,5 М растворв гексане, 6,8 мл) добавляют к перемешанной суспензии бромистого 3-метилбутил-трифенилфосфония (4,1 г) всухом тетрагидрофуране (50 мл) при-70 С, после чего добавляют раствор6-бромгексанала (1,8 г) в сухом тетрагидрофуране (6 мл). Бледную смесьоперемешивают в течение 1 ч при -70 С,после чего выпаривают в вакуумеОстаток экстрагируют с помощью простого эфира, а экстракт выпаривают дополучения бледного масла, котороеподвергают хроматографированию насиликагелевой колонке, элюируя смесью простого эфира и гексана в соотношении 1:1, с получением целевогосоединения в виде бесцветного масла.МЯМ 232/234,В. Бромистый (9-метил-деценил)трифенилфосфоний,30Раствор 9-бромо-метил-.децена(О, 9 г) и трифенилфосфина (1 5 г) вксилоле (50 мл) нагревают с цельюдефлегмации в течение 4 дней, Смесьохлаждают, верхний слой декантируют,а остаток промывают эфиром и высуши 35вают под вакуумом с получением целевого соединения в виде бледной смолы.С, Сложный метиловый эфир (1 Я,2 Я)-3-(4-формил,2-оксидобут(Е) -енил)бензойной кислоты.Это соединение получено из продукта примера 2 (стадия Е) с помощью способов, описанных в примерах 2(стадия Р) и 1 (стадия С),П. (18, 2 К)-3- 2-(2-Карбоксиэтил 45тио) -1-окси-метилпентадека(Е),5 (2), 11 (2)-триенил бензойная кислота.Указанное соединение получено изсоединений стадий В и С с помощьюспособов, описанных в примерах(1,0 г) в этаноле (40 мл) подвергают гидрированию в течение 20 мин при 56 1 Одавлении 4,22 кгс/см над окисью платины (10 мг).Катализатор отфильтровывают ифильтрат выпаривают с целью получения целевого соединения в виде бледного масла,В. Бромистый 9-метилдецилтрифенилфосфония.Раствор 1-бромо-метилдекана(1,0 г) и трифенилфосфина (1,7 г) вксилоле (50 мл) нагревают с дефлегмацией в течение 24 ч. Смесь охлажда"ют, верхний слой декантируют и остаток промывают эфиром, затем высушивают путем добавления бензола и выпаривания с целью получения целевогосоединения в виде бледной смолы.С. (18, 2 К)-3- 12-(2-Карбоксиэтилтио) -1 -окси4-ме гилпентадека(Е),5(2)-диенил бензойная кислота,Соединение получено из соединения,полученного на стадии В и сложногометилового эфира (18, 28)-3-(4-формил,2-оксидобут(Е)-енил бензойной кислоты (стадия С) при помощиспособов, описанных в примерах 1(стадия П), 3 (стадии А, В), М 1 (ББА)М-Н 1 461,П р и м е р 9. (18, 2 К)-3-12-(2 Аминокарбонилэтилтио)-1-оксипентадека - 3(Е), 5(Е)-диенил бензамид.Раствор (1 Б, 2 К)-3- 12-(2-карбоксиэтилтио)-1-оксипентадека(Е), 5(Е) -диенил 1 бензойной кислоты, сложногодиметилового эфира (50 мг), в метанольном растворе аммиака (2 мл) вьюдерживают при 50 С в герметичной бутыли в течение 3 недель. Коричневыйраствор выпаривают, а остаток очищают с помощью противофазной высококачественной жидкостной хроматографиис целью получения целевого соединения в виде бледного твердого вещества, т, пл. 83 - 84 С, МБ (ББА)М-Н 445.А. 3-Цианокоричная кислота.3-Цианобензальдегид приведен вовзаимодействие с малоновой кислотойс помощью способа, описанного в примере 2 (стадия А), с целью получения целевого продукта, т. пл, около240 С.В. 3-Аминокарбонилкоричная кислота.Раствор 3-карбоксиметилкоричнойкислоты (427 г) в диметилформамиде(4, 25 л) и водном аммиаке (удельныйвес 0,88; 8,5 л) выдерживают при ком 11 14натной температуре в течение 4 дней,подвергают концентрированию приблизительно до 11 л и затем обрабатывают с помощью льда (8 кг) и концентрированной соляной кислоты (1 л),Выпавший в осадок целевой продуктпромывают водой и высушивают, тпл260 С.С, 3-Цианокоричная кислота (альтернативный метод),Оксихлорид фосфора (746 мл) добавляют к перемешанной суспензии 3-аминокарбонилкоричной кислоты (765 г)в диметилформамиде (7,65 л), Полученный раствор нагревают при 70 -80 С н течение 50 мин, охлаждают до50 - 60 С, выливают на лед (40 л)с целью выпадения в осадок целевогосоединения, которое промывают водойои высушивают, т. пл. 242 С,Р. 3- Цианокоричный спирт,Кислоту со стадий А и С превращают в спирт с помощью процессов, описанных в примере 6 (стадия А), получив целевое соединение в виде низкоплавкого твердого вещества белогоцв ета.Е. (1 Я, 2 К)-3- 2-(2-Карбоксиэтилтио)-1-оксипентадека-З(Е), 5(2)-диенил 1 бензонитрил, сложный метиловыйэфир,Указанное соединение готовят изсоединения стадии Р с помощью процессов, описанных в примерах 2 (стадии Е и Р), 1 (стадии С и 0) и 3(стадия А).Р. (1 Б, 2 К)-3-12-(2-Карбоксиэтилтио)-1-оксипентадека-З(Е), 5(2)-диенил бензонитрил.Раствор сложного метилового эфира(200 мл) и 0,2 М раствор карбонатакалия (95 мл) перемешивают при комнатной температуре в течение 16 ч,концентрируют до 70 мл, разбавляютводой (50 мл)и промывают эфиром(50 мл). Водную фазу подкисляют дорН 3 и экстрагируют с помощью дихлорметана (Зф 50 мл). Экстракт высушиваюти выпаривают с целью получения целевого продукта в виде бледной смолы,МБ (ББА) М-Н 428,П р и м е р11. (1 Я, 2 К)-5-3(2-Карбоксиэтилтио)-1-оксипентадека 3(Е), 5(2)-диенил 1 фенил -1 Н-тетразол.Перемешанная суспензия хлористогоаммония (5 г) и азида натрия (5 г) в02256 12растворе (1 Б, 2 К)-3-12-(2-карбоксиэтилтио) -1 -оксипентадека-З(Е), 5(2)- диенил бензонитрила (780 мг) в диме 5 10 15 20 25 ЗО 35 40 45 50 55 тилформамиде (25 мл) нагревают при100 - 105 С в течение 12 ч. Темнуюсмесь отфильтровывают и фильтрат разбавляют М соляной кислотой (250 мл)и экстрагируют с помощью дихлорметана (3150 мл). Экстракт промывают водой, высушивают и выпаривают с получением темного масла, содержащего3(Е)-, 5(2)- и 3(Е)-, 5(Е)-изомерыв соотношении 30:70, Изомеры разделены с помощью препаративной противоточной высококачественной жидкостнойхроматографии с получением целевогосоединения в виде рассыпчатого твердого вещества,П р и м е р 12. А. (1 Б, 2 К)-5- 3-2- (2-Карбоксиэтилтио) -1-оксипентадека-З(Е), 5 (Е)-диенил 1 фенил 3-1 Нтетразол,Указанное соединение выделено изреакции, описанной в примере 11, МБ(ВВА) ГМ-Н 3 471,В. (1 Б, 2 К) -5-3- 2- (2-Карбоксиэтилтио)-1-оксипентадека(Е), 5 (Е)лиенил 1 Фенил 1-1 н-тетравел, натриеваясоль.Раствор продукта стадии А (273 мг)растворяют в 0,5 М растворе бикарбоната натрия (1,16 мл) и раствор подвергают сушке при отрицательной температуре с целью получения целевогосоединения в виде твердого веществабледного цвета,П р и м е р 13. (1 Б, 2 К)-3-2(2-Карбоксиэтилсульфинил)-1-оксипентадека-З(Е), 5(2)-диенилбензойная кислота..0,5 М раствора периодата натрия(1,8 мл) добавляют к перемешанномураствору (1 Б, 2 К)-3- 2-(2-карбоксиэтилтио)-1-оксипентадека-З(Е), 5(2) -диенил бензойной кислоты (372 г) в0,5 М растворе бикарбоната натрия(3,2 мл) и метанола (3,2 мл) притт0-.5 С. Смесь перемешивают в течениеО1,5 ч при 0-5 С, затем разбавляютводой, подкисляют до рН 3 и экстрагируют с помощью смеси дихлорметанаи метанола в соотношении 3:1, Экстракт высушивают и выпаривают с получением целевого соединения в видерассыпчатого твердого вещества, которое с помощью противоточной высококачественной жидкостной хроматографии содержит две диастереизомерных(1 Б, 2 Я)-3- (1,2-оксипентадека (Е), 5 (2)-диенил) бензойная кислота, сложный метиловый эфир (1,78 г), полученные аналогично примеру 4 (стадия А), растворяют в растворе 3-тиопропионитрила (0,44 г) в метаноле (5 мл) и триэтиламине (0,5 мл) в азотной атмосфере. Этому светлому раствору дают возможность отстояться50 при комнатной температуре в течение 6 ч и затем выпаривают до сухости, Полученное масло бледно-желтого цвета подвергают хроматографии на силикатной колонке, элюированной с помощью смеси простого эфира и гексана в со 55 отношении 50:50. Необходимый продукт1-1-ок сипентадек а(Е), 5 (Е) -диенилФенил -1 Н-тетраэола по примеру 13.Два диастереоизомера отведены с помощью препаративной противоточнойвысококачественной жидкостной хроматографии, МБ (ББА) М-Н 48715П р и м е р 15, (1 Я, 2 К)-5-3.2-(2"Карбоксиэтилсульфонил)-1-оксипентадека(Е), 5(Е)-диенил 1 фенил -1 Н-тетразол,Раствор персульфата калия (150 мг)в воде (0,5 мл) добавляют к перемешанному раствору (1 Б, 2 К)-5- 3-2(2-карбоксиэтилтио)-1-оксипентадека(Е), 5 (Е)-диенилфенилН-тетразола (50 мг) в 0,5 М растворе бикарбоната натрия (2 мл) и метаноле(1 мл) при 0-5 С. Смесь перемешиваютОв течение 4 ч при 0,5 С, разбавляютводой, подкисляют и экстрагируют спомощью смеси дихлорметана с метанолом в соотношении 3:1, Экстракт высушивают и выпаривают с получением целевого соединения в виде рассыпчатого твердого вещества, МЯ (ББА) М-Н503,П р и м е р ы 16 и 7. (1 Я, 2 К)- 353- 2- 2-(1 Н-Тетразол-ил)этилтио 11-оксипентадека(Е), 5(Е)-диенилбензойная кислота и 3(Е), 5(Е)-изот.мер.40 56 14зойной кислоты (1,39 г) полутаю)т в виде бесцветного масла.Этот сложный эфир (1,25 г) раствряют в тетрагидрофуране (10 мл) и добавляют 1 М водный рас.твор гидрата окиси лития (3 мл), Этот раствор перемешивают в течение всей ночи при комнатной температуре в атмосфере азота. По окончании этого периода к содержимому добавляют 1 М водный раствор гидрата окиси лития (2 мл),3 раствор подогревают до 30 С в течение 3 ч. После этого раствор выпаривают с целью удаления тетрагидрофурана, а оставшийся водный раствор доводят до рН 3 с помощью 2 М соляной кислоты. Этот раствор трижды экстрагируют с помощью простого эфира и собранные эфирные экстракты высушивают (МАМБО,) и выпаривают до почти бесцветного масла, которое медленно кристаллизуется при 0 С с получением (1 Я, 2 К) -3-2- 2-цианотил - 1-оксипентадека(Е), 5(2)-диенил бензойной кислоты. Эту свободную кислоту (500 мг) растворяют в диметилформамиде (О мл), добавляют азиц натрия (2 г) и хлорид аммония (2 г), перемешанный раствор нагревают до 120 С в течение 5,5 ч. В конце этого периода смесь разбавляют водой (30 мл) и рН раствора регулируют до 3 с помощью разбавленной соляной кислоты перед экстракцией, 5 раз с помощью простого эфира, Эфирные экстракты высушивают (МЕЯОБ) и выпаривают до получения коричневого масла. Масло растворяют в смеси метанола с водой (в соотношении 85:15) и подвергают препаративной противоточной высококачественной хроматографии на колонке с элюированием метаноловодной смеси в соотношении 85;15, содержащей 0,57. уксусной кислоты. 3(Е), 5(Е)-изомер (1 Б, 2 К) -(3-2-(2-1 Н- тетразол-ил) этилтио 1 - 1-оксипентадека(Е), 5 (Е)-диенил бензойной кислоты после первоначального элюирования подвергнут обработке болеебогатым изомером 3(Е), 5(Е). П р и м е р ы 18 - 25. Соединения формулы(СН ) СИ,получены согласно примера 3 с использованием соответствующих тиолов.П р и м е р 26, А. 3-(1,2-0 ксипентадека(Е)-енил) бензойная кислота, сложный метиловый эфир.н-Бутиловый литий (1,5 М растворв гексане, 3,3 мл) по каплям добавояют к, перемешанному раствору бромистого додецилтрифенилфосфония(50 мл) при -70 С в азотной атмосфере. Раствор глубоко оранжевогоцвета перемешивают в течение 10 минопри -70 С, затем добавляют раствор3-(2-формил,2-оксидоэтил)бензойнойкислоты,.сложного метилового эфира(1,03 г), в тетрагидрофуране (5 мл),Бледной суспензии дают возможностьнагреться до комнатной температуры,выпаривают, а остаток подвергают экстракции с помощью смеси простогоэфира и гексана в соотношении 1;1.Экстракт выпаривают, а остаток подвергают хроматографии,на силикагелевой колонке, элюируя его с помощьюсмеси простого эфира и гексана всоотношении 1:1. Целевое соединениеполучено в виде бесцветного маслаотверждаемого при охлаждении.В. Ке 1-(1 К, 28)-3-2-(2-Карбоксиэтилтио)-1-оксипентадека(Е)-енилбензойная кислота.Указанное соединение получают изпродукта стадии А примера. 3.П р и м е р ы 27 и 28. Ке 1-(1 К,28) -3-2" (2-Карбокс изтилтио) -1-ох сиундека-Э (Е), 5 (Е) -диенил бензойнаякислота и Ке 1- (1 К, 28) -3-12-карбоксиэтилтио-оксинонадека(Е), 5 (Е)диенилбензойная кислота,Указанные соединения получены изсоответствующих бромидов фосфония попримерам 1 (стадия Р), 3 (стадии Аи В).П р и м е р 29. А. Бромистый(50 мл) нагревают с целью дефлегмации в течение 8 ч. Раствор выпаривают и остаток промывают простым эфиром с получением целевого соединенияв виде гигроскопического белого кристаллического твердого вещества, которое высушивают путем добавлениябензола и повторного выпаривания.10 В. 2-(11-Фенил-ундеценилокси) -тетрагидропиран,н-Бутиловый литий (1,6 М растворв гексане, 20 мл) добавляют к перемешанному раствору продукта (11,0 г)стадии А в сухом тетрагидрофуранеопри -70 С в азотной среде, Оранжевый раствор перемешивают в течение30 мин при -70 С, после чего добавляют к содержимому раствор Э-фенилпропиональдегида (2,75 г) в тетрагидрофуране (7 мл), Бледному раствору дают возможность нагреться до комнатной температуры и затем выпаривают, Остаток экстрагируют с помо 2 Б щью простого эфира и экстракт вновьвыпаривают, а остаток подвергают хроматографии на силикагелевой колонке,элюируя ее с помощью смеси простогоэфира и гексана в соотношении 1:1 сЗп целью получения целевого соединенияв виде бледного масла.С. 11-Фенил-ундеценол.Раствор продукта из стадии В(8,7 г) в тетрагидрофуране (150 мл)35и 2 М соляной кислоты перемешиваютпри комнатной температуре в течение4 ч. Смесь нейтрализуют растворомбикарбоната натрия и экстрагируют спомощью дихлорметана. Выпаривание экстракта и хроматография остатка насиликагелевой колонке, элюируя с по,мощью смеси простого эфира и гексана в соотношении 1:1 с целью удаления исходного материала, а затем с45помощью простого эфира, дают целевойпродукт в виде бледного масла.Р. 11-Фенил-ундеценолтозилат.4-Толуолсульфонилхлорид (1,3 г)добавляют по частям к перемешанномураствору продукта стадии С (1,5 г)50Ов пиридине при 0-5 С. Смесь перемеошивают в течение 16 ч при 0-5 С, после чего сливают в ледяную солянуюкислоту и экстрагируют простым эфиром. Экстракт промывают растворамибикарбоната натрия и хлористого натрия, высушивают и выпаривают с целью получения целевого соединения ввиде бледного масла,
СмотретьЗаявка
3777634, 01.08.1984
Лилли Индастриз Лимитед
СТЕФЕН РИЧАРД БЕЙКЕР, УИЛЬЯМ БОФФИ ДЖЭМИСОН, АЛЕК ТОДД
МПК / Метки
МПК: A61K 31/10, A61K 31/19, A61P 37/08, C07C 317/44, C07C 323/56, C07C 323/60, C07D 257/06
Метки: органических, серусодержащих, соединений, солей
Опубликовано: 07.06.1988
Код ссылки
<a href="https://patents.su/19-1402256-sposob-polucheniya-serusoderzhashhikh-organicheskikh-soedinenijj-ili-ikh-solejj.html" target="_blank" rel="follow" title="База патентов СССР">Способ получения серусодержащих органических соединений или их солей</a>
Способ получения производных флавана или их солей

Номер патента: 1072805
Опубликовано: 07.02.1984
Авторы: Гарольд, Денис, Джон, Дэвид
МПК: A61K 31/352, A61P 31/12, C07D 311/60
Метки: производных, солей, флавана
...пыли и 0,8 г хлористой ртути) в ноде (30 мл), и смесь перемешивают при 45 С в течение 45 мин. Полученный цинк отделяют декантиронанием от водной жидкости, перемешивают со 150 мл уксусной кислоты и 30 фильтруют, Цинк дополнительно промывают уксусной кислотой, объединенные промывочные воды и фильтрат разбавляют водой и экстрагируют толуолом. Экстракт промывают водой, 35 сушат под сульфатом магния, фильтруют и выпаривают. Остаток хроматографируют на нейтральной окиси алюминия, упаривают первую фракцию. В результате получают кристаллический 40 4 -(И,И-диметиламино)-флаван, который подвергают перекристаллизации из метилового спирта и затем из петроЛейного эфира 60-80 С получая 0,7 продукта с т.пл. 77-78 С.6 П р и м е р 39. Синтез 4...
Способ получения метилового фиолетового окисления диметиланилина с помощью соединений железа

Номер патента: 15429
Опубликовано: 31.05.1930
Автор: Белоцерковский
МПК: C09B 57/00
Метки: диметиланилина, железа, метилового, окисления, помощью, соединений, фиолетового
...(солей или свежеосажденных гидратов окислов) или их смесей с обычными разбавителями (поваренной солью, песком илит. и.) прибавляют диметиланилин и нагревают при доступе воздуха под атмосферным или повышенным давленйем, после чего либо раство. ряют смесь в разбавленной соляной кислоте и выделяют краситель обычными способами, либо экстрагируют краситель из сухой смеси ацетоном. ПЯТЙ 11 Т ПЯ пособа получения мет В технике производства красок известен процесс окисления смеси диметиланилина с дифениламином в фиолетовый краситель, происходящий при действии на нее хлорного железа и извлечения из реакционной смеси спиртом трифенил метановых красителей.В предлагаемом способе получениеметилового фиолетового осуществляетсяприбавлением...
Способ получения производных

Номер патента: 416944
Опубликовано: 25.02.1974
Авторы: Адольф, Иностранна, Йозеф, Рольф, Руди
МПК: C07D 239/95
Метки: производных
...в вакууме досуха. Остаток поглощают в разбавленной соляной кислоте и полученный таким образом раствор фильтруют до осветления, Затем водный раствор соляной кислоты подщелачивают содовым раствором и выделяющийся в виде масла продукт реакции поглощают уксусным эфиром. После сушки над поташом при введении сухого хлористого водорода в уксусноэфирный раствор получают гидрохлорид 3- у-диэтиламино-Р- (3,4,5-триметоксибензокси) -пропил -6,7,8-триметокси- (1 Н,ЗН) - хин азолин-тион-она в виде бесцветных игл,т. пл. 154 - 156 С; Выход 43 г (68,5 от теоретического) . 11. Применяемый в качестве исходного про дукта 3-(у-диэтиламинооксипропил) -6,7,8 триметокси-(1 Н,ЗН)-хиназолин-тион - он можно получить следующим образом, 28,3 г (0,1 моль)...
Способ получения 16, 17-ацетальзамещенных прегнан-21овой кислоты в виде их стереоизомеров

Номер патента: 1839673
Опубликовано: 30.12.1993
Авторы: "пьер, Бенгт, Бро, Поль, Ральф, Ян
МПК: C07J 5/00
Метки: 17-ацетальзамещенных, виде, кислоты, прегнан-21овой, стереоизомеров
...равна 98,9(. Температура плавления 192 - 195 С. ( ар =+66,4(с = 0,256; СН 2 С 2), Молекулярный вес 522,П р и м е р 5. Раствор 0.45 ацетата медив 150 мл метанола добавляют к раствору 1,0 г (228)-16 а, 17 а-бутилидендиокси-ба, 9 а -фтор11, 21-диоксипрегнан,4-диен,20-диона в 150 мл метанола и подвергают взаимодействию. Образовавшийся продукт выделяют так, как описано в примере 1, Этот продукт растворяют в 15 мл изопропанола и выпаривают. Эту процедуру повторяют дважды, в результате чего получают 1,2 г (22 й)-16 а, 17 а -бутилидендиокси-б а, 9 а -дифтор1 -окси,20-диоксопрегнан,4- диен-ал-пропил полуацеталя,Этот альдегид подвергают взаимодейг ствию в условиях, описанных в примере 1, заменив метанол изопропанолом, Неочищенный продукт...
Способ получения оптически-активных производных 9-дезокси проста-5, 910, 13-триеновых кислот, или их рацематов

Номер патента: 650500
Опубликовано: 28.02.1979
Авторы: Джианфредерико, Кармело, Пьетро
МПК: A61K 31/5575, C07C 57/26
Метки: 13-триеновых, 9-дезокси, кислот, оптически-активных, производных, проста-5, рацематов
...К 5, Кб, и то нием соединени н,г 5 с (сн 1 сн В 6 7+ уогде К, У", К 5, К 6 К, представляет с алкил С 1 - С, или С - С 6 (158-ол) и Х 17 б и и то же, что и выше, бой атом водорода или алкенильную группусоединения формулы 40 где К, У", К 5, К 6 и иК 4 представляет соалкил С, - СилиС 2 - С, (15 К-ол) свосстановителем иливом Гриньяра, в завинений, которые нужно6) Разделение 155пример, хроматографхроматографической7) Этерификацияили смеси 155-ола исоединения формулы по же, что и выше, бой атом водородаалкенильную групомощью обр або по реакции с реа симости от тех со получить.-ола от 15 К-ола, ированием, лучше колонке;155-ола или 15 К 15 К-ола с получен ХЧ тина СНСООВ,С (СН) СН гг Б кф ягде К, У", К Один из К 3 водорода или ную группуже, что и...
Предыдущий патент: Способ получения n-изопропил-n -о карбометоксифенилсульфамида
Следующий патент: Способ получения 3 -цианоморфолинопроизводного доксорубицина
Случайный патент: Пневматическое пусковое устройство для приводов высоковольтных воздушных выключателей