Способ получения три-н-бутилтритиофосфата
Похожие патенты | МПК / Метки | Текст | Заявка | Код ссылки




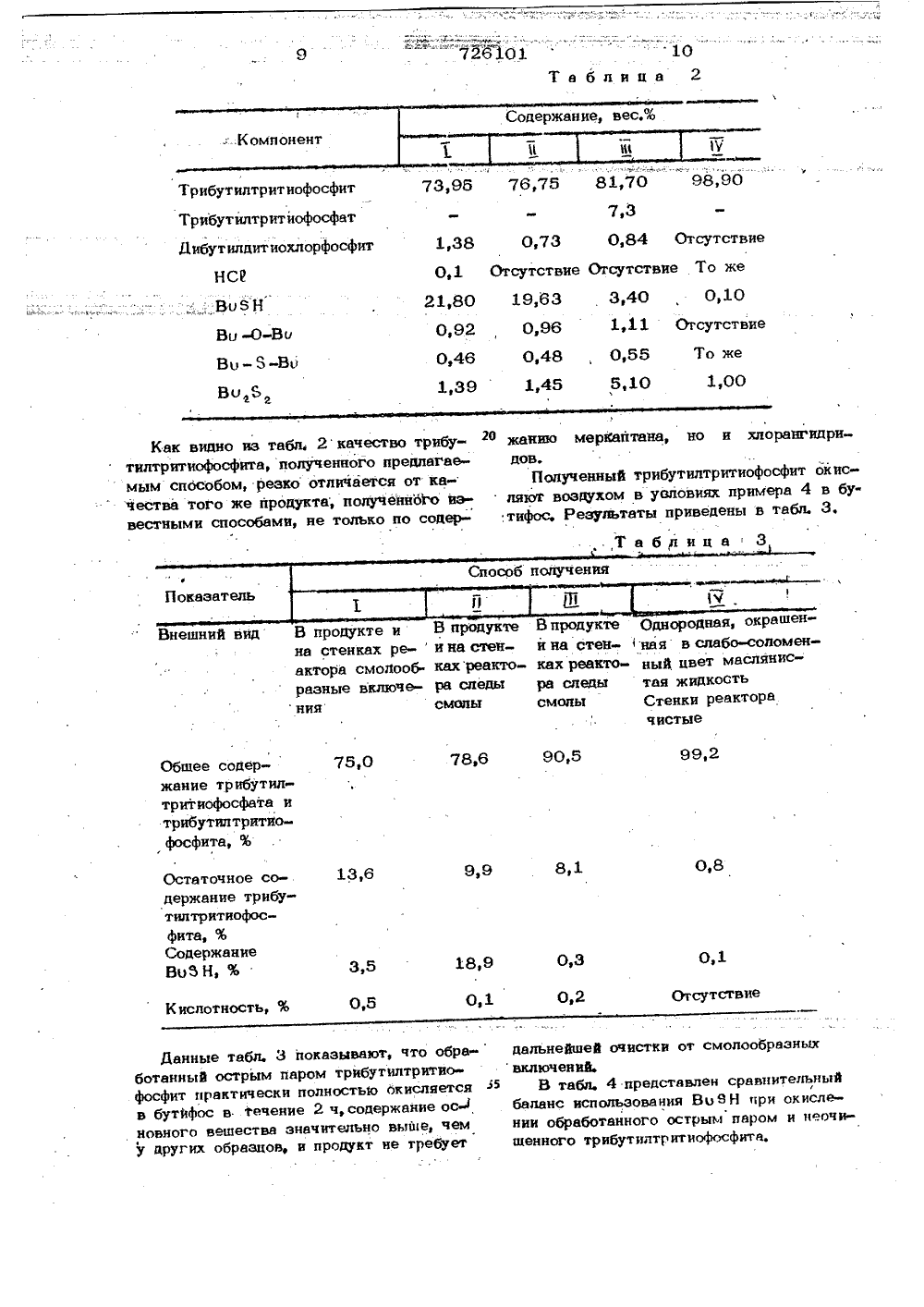

Текст
соелине ая Гасударственный квмнте СССР23) Приоритет етень 107.04,80 53) УДК 547.26 11 18 07(088 е 8 елам изобретений н открытий Опубликовано 05.04.80, Бюлл опубликования описан И, М. Мильгот инин, Р А, П. В,Алпатова, С, Н.Комова, арфенов и В. Н, Троицк 72) Авторы изобретен аявитель,Иля ускорения процесса фосфит окисляступенчато; вначале в течение 2-6 ч духом при 230 С, а затем 50%-ным твором перекиси водорода при 230 С Я.Выход бутифоса, а также состав при- Й ей не указаны.Способ мало применим в промышленти как из-за опасности работы с контрированной перекисью водорода, так следствие удорожания процесса в резуль. е использования последней.звестен способ получения бутифоса псодом 95,4%, в котором получение ита осушествляют при 38149 С во ние 6-8 ч с последуюшей обработкой ционной массы основаниями, напримерййЭ ином, карбонатом или бикарбонатом ия или диметиланилином, для связывахлористого водорода (НС 1 ) и побочпродуктов, фосфит окисляют кислорс- воздуха при 110 С в течение 1031 отности про- одоЦизбыток оэдухом Ц. Известен способ поимодействием РСРольном соотношенн- 4Изобретение относится к химии фосфор- органических соединений, а именно к спо- ют собу получения три н-бутилтритиофосфата . воз (бутифоса), широко применяемого в сель-. рас ском хозяйстве для дефолиации хлопчатни5 ка, а также в качестве присадки к смазочным маслам,месИзвестен способ получения бутифоса путем взаимодействия треххлористого фосфора нос (РСс ) с бутилмеркаптаном (ВобН), взятых цен 1 ввмольном соотношении 1:(3-3,5), при тем- и в пературе до 150 оС в течение 6,5 ч е . тат последующим окислением полученного фосфита кислородом воздуха при 110 С. ИВыход бутифоса 82% конверсия ВоЗН с вь 90%, фосфДля освобождения от кисл тече дукт обрабатывают горячей в реак ВоЗН удаляют продувкой в пириднатр учения бутифоса ния с ВоЭН, взятых ных 1;3,2, при 100- дом18 ч И-н-БУТИЛТРИТИОФОСФАТА101 40 гфосфору. 3 726Обработка основаниями удорожает про-,цесс и приводит к образованию значительного количества одориромнных стоков.Большаядлительйость"окйсления требует 1 " установки оборудования большой емкости.Снятие кислотности основаниями не обеспечивает удаления меркаптайа","поэтомуон теряется при окислении.Использование в производстве бутифо"са сйособа его очистки путем обработки 1 Оперегретым водяным паром Я позволяет обеспечить повышение качестм товарного продукта, исключить кислотность товарной формы и тем самым повысить стабильность препаративной формы при хра- . 15ненни в металлической таре, но не исключает значительнуо-пожаро 6 йасйость приокислении фосфита, поскольку процесс ве- -дется в избытке кйслорода воздуха с одно= временной отдувкой меркаптана и при этом 20образуется" взрывоопасная смесь меркаптана е кислородом воздуха.Ближайшим по технической сущностии достигаемым результатам к изобретению является способ получения бутифоса=йутем"взаимодействия РСЖ с избыткомВо 1 Н при 135-240 С с йоследующим "- -" - -окислением наученного трибутйлтритио-,фосфита молекулярным кислородом иликислородом воздуха при вышеуказанной " "температуре, в котором как трибутилтритиофосфит, так и бутифос промывают во-дой, с последующей обработкой "газовойфазы, содержащей НСФ и ВоЬ Н раствором едкого патра Я.35Применение водной промывки позволя етснять кислотность, однако не обеспечивает полное удаление и рецикл избытка.вводймого ВОЬН, а также удаление продуктов его окисления - дисульфидов - изтехнического бутифоса.Использование едкого натра для нейтрализапии всего выделяющегося НС 1 "нецелесообразно по экономическим соображениям.45 окислением три-и-бутилтритиофосфита вбутифос обрабатывают острым паром.Обработку реакционной массы острымпаром желательно проводить при темпераотуре 100-115 С и атмосферном давленииили при температуре 60-100 С в ва-,кууме 200-100 мм рт,ст. и времени кон;такта 10-80 мин.Установлено, что три-н-бутилтритиофосфит при обработке острым паром, лучше,в динамических условиях, за времяконтакта до 80 мин при 100-120 С стабилен и не гидролиэуется. Установленотакже, что применение острого пара обеспечивает практически полное удалениеВОЙН,Конденсация дистиллята, состоящего восновном из водяного пара и меркаптана,обеспечивается при использомнии воды вкачестве охлаждающего агента.Очистка три-н-бутилтритиофосфита острым паром легко осуществляется в динамических условиях в насадочной колонне,Расход пара, 0,5-0,6 кг/кг возвращаемого Во 5 Н. Водный конденсат практически не содержит органических примесей ииспользуется на стадии поглощения НС 1.Очищенный в перегонной колонне три-н-бутилтритиофосфит далее при времениконтакта до 2 ч и температуре 110 о120 С нацело окисляется кислородом воздуха в бутифос,Таким образом, синтез три-н-.бутилтритиофосфита по обычной схеме, но с последующей обработкой его острым паром,обеспечивает полный возврат Во 5 Н в цикли очистку фосфита от ингибируюших процесс окисления примесей, вследствие чегоокисление протекает в более короткое время с получением целевого продукта высокого качестм. Во всех примерах выходи содержание основного вещества даны по П р и м е р 1. В прибор .для отгонкис острым водяным паром помещают 100 гЦелью изобретения является упроще. ние процесса и повышение выхода и чистоты целевого продукта.Поставленная цель достигается спосо Обом получения бутнфоса путем взаимодей ствия РС 1 с Во 5 Н при -нагревании споследующей обработкой реакционной масмсь острым паром и окислением очищенно го три-н-бутилтритиофосфнта кислородомвозд уха еОтличительным признаком способа является то, что реакционную массу перед трибутилтритиофосфита (пв 1,5305;4 1,0420; т, кип. 150-152 С/2 мм рт.ст.), подогревают куб ро 100-105 С и через фосфит барботируют острый водя ной пар, пары конденсируют в холодильнике и собирают в приемник. Через 40- 60 мин дистиллят не содержит органической фазы н имеет слабый запах. Кубовая жидкость расслаивается, нейтральную водную фазу отделяют, а органическую отпаривают от остаточной влаги при 120"С/ /5 мм рт.ст.5 726Получают 100 г продукта, п 1,5305;д 1,0421;т,кип. 150-152"С/2 ммрт,ст,Таким образом, обработка трибутилтритиофосфита острым паром не вызывает 5его гидролиза.П р и м е р 2, В реактор, снабженный мешалкой, обратным холодильником,термометром и системой поглощения кислых паров, помещают 68,75 г(0,5 моль) 1 ОРС 0 и при перемешимнии приливают182 г (2 моль) 99%-ного ВиЬН, нагремют за 10 мин до 75 С и выдерживаютпри 75-80 С в течение 80-90 мин, затем температуру повышают до 125 оС и 15выдержимют 2 ч при 125-135 С. Послеохлаждения получают 190 г техничес-кого трибутилтритиофосфита, а от воднойфазы системы поглощения отделяют 2,03 гВи 5 Н. гоВыход 99,5%, содержание основноговещества 78%.П р и м е р 3, В нагреваемую до105-120 С электроспиралью стекляннуюколонку диаметром 45 мм и высотой25600 мм с насыпанными на высоту 350400 мм кольцами Рашига диаметром3 мм сверху из кацельной воронки равномерно по каплям вводят 100 г/ч технического трибутилтритиофосфита, полученного в примере 2,Одновременно в нижнюю часть колоннывводят острый водяной пар и отгоняют спаром летучие примеси через холодйльникв приемник конденсата, где он расслаим- З 5ется на водную и органическую фазу, Вкубе колонны также получают две фазы:нижнюю, водную, слегка кислую, и верхнюю, органическую, маслянистую.40При установившемся режиме отпаркитемпература верха колонны 88-92 С,средоней части 105-110 С, куба 80-90 СИз 100 г/ч вводимого техническогофосфита получают в приемнике дистиллята21,6-21,8 г/ч ВоВН, и 25-35 г/ч воды.45В кубе колонны получают 20-50 г/ч вод-нокислотной фазы и 78,1-78,0 г/ч очищенного трибутилтритиофосфита. Послеосушки" в техническом вакууме при 100 :получают фосфит, о 1,5305;1,0421.ю, до ЮоВыход 99,5%, содержащие основного вешества 99,8%.Водный конденсат в приемнике имеетнейтральную реакцию, кислотность водной 101офазы в кубе колонны 0,1-3% в пересчете на соляную кислоту.П р и м е р 4. В стеклянную колонку диаметром 30 мм и высотой 250 мм,снабженную пористой пластинкой, обратнымхолодильником, термометром и трубкойдля подвода воздуха, загружают 100 гочищенного фосфита, полученного в примере 3 или 1, холодильник присоединяюткловушке охлаждаемой смесью сухого льдас ацетоном, нагревают до 100 С и пропускают 32-35 л/ч воздуха, при повышении температуры выше 120 С колонкуслегка охлаждают и проводят окислениев течение 2 ч при 110 3.20 С.Получают 105,4 г бутифоса, содержащего 0,7% трибутилтритиофосфита, Общеесодержание.фосфата и фосфита 99,8%.В ловушке отсутствует конденсат. Выход на стадии окисления близок к количественному.При увеличении времени пропусканиявоздуха до 4 ч получают продукт, содержащий 0,2-0,3% трибутилтритиофосфита,П р и м е р 5.Из 68,75 г(0,5 моль)РСЮ и 186 г (2 моль) 97%-ного ВоЗНв условиях примера 2 получают 200,1 гнеочищенного технического фосфита и вохлаждаемой до -70 С ловушке 2 г ВирН.Выход 99,5%, содержание основного вещества 73,95%.П р и м е р 6. В условиях примера3 при подаче 70 г/ч технического фосфита, полученного в примере 5, получают17,5 г/ч 90,0%-ного Ви 8 Н и 52,3 г/чтрибутилтритиофосфита, содержащего98,9% основного вещества,Выход 99%.Состав отогнанного Воб Н, вес.%фХлорбутил (Во С 6) СледыВобН 90,0Дибутиловый афир(Во Ья ) 4,6Указанные примеси вносятся с исходным ВиЗНВ табл. 1 приведен сравнительный средний состав Технического и очищенного острым паром трибутилтритиофосфита, лолученного при использомнии 97%-ногоВо 3 Н ( моль ное соотношение РСЮ ; Во б Н1:4).8Таблица 1 726 101 Содержание, вес.%, в фосфите К омпонент техническом очищенном Трибутилтритиофосфитй-Дибутилдитиохлорфосфит 73,95 98,90 1,38 Отсутствие НС 1 0,1 То же В(.) 5 НВ(.) -0-ВоВо -3 -В(.)В(.) ) 21,8 0,1 ОтсутствиеТо же 0,92 0,46 1,00 П р и м е р 7. Из 68,75 г(0,5 моль), и 300 г (2 моль) возвратного (60%-ноРСР и 200 г (2 моль) ВИЛЬН-сырца сого) ВибН, натревают 15 мин до 75 оС,става, вес.%: ВобН 90, ВоС 1 2,03, . выдерживают при атой температуре 1,5 ч,Во-В 1,2, Во-Во 2,6, В(,) б затем температуру повышают до 120 С4,1, Н 0 0,07, в условиях примера 2аи выдерживают при этой температуре 2 ч,получают 212,6 г технического фосфита, Получают 297,5 г фосфита, содержащеВыход 98,7%, содержание основного25 го 49,3% основного вещества.вещества 69,2%. Выход 99%.В ловушке конденсируется 1,2 г В(,(5 Н П р и м е р 9. 200 г технического100 г, полученного фосфита обрабаты- фосфита, полученного в примере 8, обраовают острым паром для удаления летучих батывают острым паром при 90-120 С.примесей в вакууме до 200 мм рт.ст, и ЗО В сборнике дистиллята получают дветемпературе куба 65-90 С. фазы: верхнюю-технический ВоВН с соОтпарку ведутдо прекращения увели- путствующими ему, примесями (99,7 г) ичения объема органической фазы в прием- .нижнюю - вода,нике дистиллята. В кубе после отпарки также обраэуетПолучают в отгоне 30,1 г органичес ся две фазы: верхняя - очищенный фосфиткой фазы и 30 г воды,в кубе также две (100 г), содержащий 98,6% трибутилтрифаэы; верхняя-очищенный трибутилтритио- тиофосфита, нижняя - водно-кислотная фаза,фосфит в (69,3 г) и нижняя - водно-кис г очищенного фосфита окисляют кислотная фаза (23 г). лородом воздуха в течение 2 ч при 100 о50 г очищенного фосфита, содержаще О 120 СПолучают 52,6 г техническогого 97% основного вещества, помешают в бутифоса с общим содержанием трибутилреактор колонного типа с пористым дном тритиофосфита и трибутилтритиофосфатаи окисляют кислородом воздуха в течение .98,6% и остаточным содержанием трибу 1,5-2 ч при 110-120 оС. тилтритиофосфита 0,95%.Получают 52,7 г бутифоса с общим 4 В табл, 2 указано качество трибутилсодержанием трибутиптритисфосфете и три- тритиофосфите понученного иевестнымбутиптритисфосфите 97,2(б и остаточным способом (91 11 - технического (1 ),прсодержанием фосфита 0,85%. мытото горячей водой ( и ) и промытогоП р и м е р 8. В реактор с мещал- горячей водой с последующей отдувкойкой, обратным холодильником и термомет-М . воздухом (1 й )-предлагаемымспособомром загружают 68,75 г (0,5 моль) РС 0 обработанного острым паром к( ).,1 Отсутствие Отсутств То ж Во 8 0,1 2 0,9 6 0,4 Ви -0 0,55 5,10 0 ж,39 1,45 0 рангидритабл. 2 качество трибуа, полученного предлагаерезко отличается от капродукта; полученно о иэобами, не только по содер 0 жанию а, но бутил та дов,ПоллякуГтифос,итиофосфит примера 4 ны в табл. окисенн Словиях ривед о зультат Внешний вид В продукте и В продукте на стенках ре-н на стенактора смопооб- кахреакто разные включе- ра следы смол 2 О,75,0 Общее содержание трибутилтритиофосфата и трибутилтритнофосфита, %Остаточное содержание трибу- тнлтритиофосфита, %Содержание ВобН, % 8,1 9,9 0,3 8,9 0,2 5 Кислотность, % стви дальнейшей очисткивключений.В табл. 4 предстбаланс использова ниянии обработанного осщенного трибутилтрит обра-3 показываюпаром трибутки полностьюение 2 ч,содева значительнов, и продукт т смолообр илтритиокисляет Л ание осыше, чемтребует ьный /ислееочивлен сравнитеВо 8 Я при окрым паром ииофосфита,Как видно из тилтритиофосфит мым способом,честна того же вестными сносДанные табл.ботанный остры фосфит практичес в бутйфос в. те новного вешесту других образц 10 Та блица 21,11 Отсутств и на стен- ная в слабо-соломках реакто- ный цвет маслянисра следытая жидкостьсмолы Стенки реакторачистыеЖ"бработанный острым парбм с О 0,75 0,73 0,0 9 4 23 еочищенный фосфи 5 3,0 3 3,0 2 94 3,5,2 2,94 0 93,2 98,0 а, реакционную масобрабатывают острым ча юострым п куу0- ин. Редактор Т. Соста анова Техреель М. КрасновскаяО. Андрейко Корректо М Синицк 598/21" Тирйй 495 Пойписное ЦНИИПИ Государственного комитета СССР по делам изо и открытий 113035, Москва, аушская наб д. 4/5 Филиал ПЛП фПатент", г, Ужгород, ул. Проектная, Заказ бетений Ж 35,Р Предлагаемый споСоб получений бути-.фоса не взрывоопасен, так как фосфит, подаваемый на окисление, не содержитВо 5 Н,.позволяет испольббйать техйИческий Во 8 Н,5содержащий 60-90% основного вещества,и тем самым исключить сложйую ректификацию ВобН, что упрощает процесс. Выход целевого продукта достигает 98%,чистота его повышается. 10Формула изобретен ия1 Способ получения три-н-бутилтритио 85фосфата"путем взаймодейСтвйя треххлористого фосфора с бутилмеркаптаном при на-гревайии с последующйм ойв:ленкам получейного"три-н-бутилтритиофоЯйта кислоро- дом воздуха при нагревании, о т л и - Оч а ю щ и й с я "тем,"что, с целью упрощения процесса и повышения выхода и чйототы целевого продуктсу перед окислениемпаром. -2. Способ по п. 1, о т л ищ и й с я тем, что обработкуром ведут при атмосферном давлении итемпературе 100-115 С или в ва м200-100 мм рт,ст. и температуре 6о100 С и времени контакта 10-80 мИсточники инФормациифпринятые во внимание при акспертиз1. Патент США Ж 2943107,кл, 260-401, опублик. 1960.2, Патент США Ж 3136807,кл. 260-461, опублик. 1964.3, Патент США М 3174989,кл. 260-461, опублик, 1965.4, Авторское свидетельство СССРЖ 374323, кл. С 07 Р 9/17, 1971.5. Патент США М 3885002,кл.: 260-972, опублик, 1975 (протот
СмотретьЗаявка
2539619, 09.11.1977
ПРЕДПРИЯТИЕ ПЯ А-7411
ВЕРШИНИН ПЕТР ВАСИЛЬЕВИЧ, АЛПАТОВА РИММА ИВАНОВНА, КОМОВА СВЕТЛАНА НИКОЛАЕВНА, МИЛЬГОТИН ИОСИФ МЕЕРОВИЧ, ПАРФЕНОВ АЛЕКСАНДР ИВАНОВИЧ, ТРОИЦКИЙ ВЛАДИМИР НИКОЛАЕВИЧ
МПК / Метки
МПК: C07F 9/165
Метки: три-н-бутилтритиофосфата
Опубликовано: 05.04.1980
Код ссылки
<a href="https://patents.su/6-726101-sposob-polucheniya-tri-n-butiltritiofosfata.html" target="_blank" rel="follow" title="База патентов СССР">Способ получения три-н-бутилтритиофосфата</a>
Способ количественного определения паров анилина в воздухе

Номер патента: 826221
Опубликовано: 30.04.1981
Автор: Стенцель
МПК: G01N 21/78
Метки: анилина, воздухе, количественного, паров
...жидкость сохраняетстабильность своих параметров 1 мес.Воспроиэводимость определения концент. раций анилина в воздухе не ниже 1,5. Среднестатическая ошибка определения для серии опытов не превышает 1,5, АП р и м е р 1. Обоснование интервалов концентраций бромфенолового синего.Приготавливают в колбах чувствитель"ные жидкости, содержащие соответст венно 0,001 т 0,0025 р 0,005; 0,01 р 0,025 и 0,05 бромфенолового синег(БФС), добавляют 1-ный раствор НС 1, в весовом соотношении 20:1. Из каждой колбй отбирают по 25 мл смеси, добав-. ляют соответственно 0,02 и 0,2 мл 0,1-ного спиртового раствора анилина и перемешивают, Через 5 мин колориметрируют на синем светофильтре.Результаты определейия анилина приведены в табл, 4. П р и м е р 2....
Способ количественного определения паров анилина в воздухе

Номер патента: 443311
Опубликовано: 15.09.1974
Авторы: Бовкун, Буковский, Воронова, Псалтыра
МПК: G01N 31/22
Метки: анилина, воздухе, количественного, паров
...из расчета на 100 г фарка 0,15 г гексанитроцерата а персульфата калия и затем сушить в течение 2 - 2,5 час при комнатной температуре,П р и м е р. 100 г фарфорового порошка с диаметром зерен 0,15 - 0,4 мм заливают 300 мл разбавленной соляной кислоты (1: 1), нагревают на кипящей водяной бане в течение 3 час, промывают дистиллированной водой до отрицательной реакции на ион хлора. Затем тот же порошок заливают 300 мл разбавленной азотной кислоты (1: 1) и снова нагревают на кипящей водяной бане в течение 3 час, промывают дистиллированной водой до отрицательной реакции на нитрат-ион и высушивают при 250 С в течение 2 час. 100 г высушенного фарфорового порошка обрабатывают 30 мл реактивного раствора, который готовят следующим образом: к 100...
Способ количественного определения паров анилина в воздухе

Номер патента: 792118
Опубликовано: 30.12.1980
Авторы: Зотова, Ковалев, Кравец, Стенцель
МПК: G01N 21/78
Метки: анилина, воздухе, количественного, паров
...и 35-ный раствор едкого калия,40 Таблица 2 Оптическая плотность раствора при добавлении1-го раствора анилина и 10 мл 35-го КОН, мл 0 0,05 0,1 0,15 0,20 0,250 0,035 0,057 0,085 0,154 0,192 0,04 0,060 0,089 0,153 0,196 0,039 0,060 0,088 0,150о 0,038 0,059 0,0870,152 0,193 0,1940 Среднее цию анилина в воздухе по концентрация анилина врастворе по калибровочномуграфику фотоэлектрическогоколориметра;объем пропущенной черезпоглотительный прибор анилиновоздушной смеси;поправочный коэффициент; В мерные колбы емкостью 50 млвливают с помощью пипетки 25 мл(точно) чувствительного раствора и добавляют соответственно 0,05, 0,1,0,15,0,2 и 0,25 мл 1-го раствораанилина. После каждого добавленияраствора анилина сразу же в колбувливают 10 мп 35-го...
Способ определения паров анилина в воздухе

Номер патента: 857808
Опубликовано: 23.08.1981
Авторы: Бахмет, Бикулов, Кавизин, Миргородский, Сагайдачный, Стенцель
МПК: G01N 21/78
Метки: анилина, воздухе, паров
...концентрации, равные 5-10 ПДАБ в ледяной уксусной кислоте.Используют сухие индикаторные ленты, пропитанные раствором, содержащим 5 ПДАБ в ледяной уксусной кислоте, на лицевую поверхность которых нанесены соответственно: 0,1 0,15;0,200,25 и 0,30 растворы бромфенолового синего индикатора. Перед анализом ленты смачивают до влажного состояния 65-ным раствором уксусной кислоты.Результаты использования растворов приведены в табл, 2. Иэ,табл, 2 видно, что наибольшая чувствительность газоанализатора соответствует содержанию бромфеноловогоФсинего индикатора в растворе, равному 0,2-0,25.Используют сухие ийдикаторные ленты, пропитанные раствором, содержащим 5 ПДАБ в ледяной уксусной 60 кислоте, на лицевую поверхность которых нанесен 0,2-ный...
Способ получения индикаторной массы для определения паров воды в воздухе

Номер патента: 1422147
Опубликовано: 07.09.1988
Авторы: Гуськова, Классовская, Кошовский, Кушнир, Поченкова
МПК: G01N 31/22
Метки: воды, воздухе, индикаторной, массы, паров
...при 500-52 ФС в течение 1,5-2,0 ч, обработанном раствором селена, и выдержанная после0приготовления нри 30-35 С в течение 2-3 сут, по чувствительности, контрастности перехода окрасок и четкости раздела на границе непрореагировавшего и прореагировавшего с парами воды слоев значительно превосходит известную индикаторную массу.Предлагаемый способ получения индикаторной массы для определения паров воды в воздухе может быть использован в производстве индикаторных трубок, предназначенных для быстрого анализа воздуха непосредственно в подземных условиях.Формула изобретенияСпособ получения индикаторной массы для определения паров воды в воздухе, состоящей из носителя с размерои частиц 0,3-0,5 мм, о 6 работанного раствором селена в...
Предыдущий патент: Способ получения трис-(хлоралкил) фосфатов с разноименными радикалами
Следующий патент: Способ получения диалкиловых эфиров изотиоцианатотиофосфорной кислоты
Случайный патент: Устройство для измельчения и термообработки сыпучих материалов